by Kim Askey, [email protected], 865.576.2841
A team of scientists led by the US Department of Energy’s (DOE’s) Oak Ridge National Laboratory (ORNL) and the Georgia Institute of Technology (Georgia Tech) is using supercomputing and revolutionary deep learning tools to predict the structures and roles of thousands of proteins with unknown functions.
Their deep learning-driven approaches infer protein structure and function from DNA sequences, accelerating new discoveries that could inform advances in biotechnology, biosecurity, bioenergy, and solutions for environmental pollution and climate change.
Researchers are using the Summit supercomputer at ORNL and tools developed by Google’s DeepMind and Georgia Tech to speed the accurate identification of protein structures and functions across the entire genomes of organisms. The team recently published details of the high-performance computing (HPC) toolkit and its deployment on Summit.
These powerful computational tools are a significant leap toward resolving a grand challenge in biology: translating genetic code into meaningful functions.
Proteins are a key component of solving this challenge. They are also central to resolving many scientific questions about the health of humans, ecosystems and the planet. As the workhorses of the cell, proteins drive nearly every process necessary for life—from metabolism to immune defense to communication between cells.
“Structure determines function” is the adage when it comes to proteins; their complex 3D shapes guide how they interact with other proteins to do the work of the cell. Understanding a protein’s structure and function based on lengthy strings of nucleotides—written as the letters A, C, T, and G—that make up DNA has long been a bottleneck in the life sciences as researchers relied on educated guesses and painstaking laboratory experiments to validate structures.
With advances in DNA sequencing technology, data are available for about 350 million protein sequences—a number that continues to climb. Because of the need for extensive experimental work to determine three dimensional structures, scientists have only solved the structures for about 170,000 of those proteins. This is a tremendous gap.
“We’re now dealing with the amount of data that astrophysicists deal with, all because of the genome sequencing revolution,” said ORNL researcher Ada Sedova. “We want to be able to use high-performance computing to take that sequencing data and come up with useful inferences to narrow the field for experiments. We want to quickly answer questions such as ‘what does this protein do, and how does it affect the cell? How can we harness proteins to achieve goals such as making needed chemicals, medicines and sustainable fuels, or to engineer organisms that can help mitigate the effects of climate change?’”
The research team is focusing on organisms critical to DOE missions. They have modeled the full proteomes—all the proteins coded in an organism’s genome—for four microbes, each with approximately 5,000 proteins. Two of these microbes have been found to generate important materials for manufacturing plastics. The other two are known to break down and transform metals. The structural data can inform new advances in synthetic biology and strategies to reduce the spread of contaminants such as mercury in the environment.
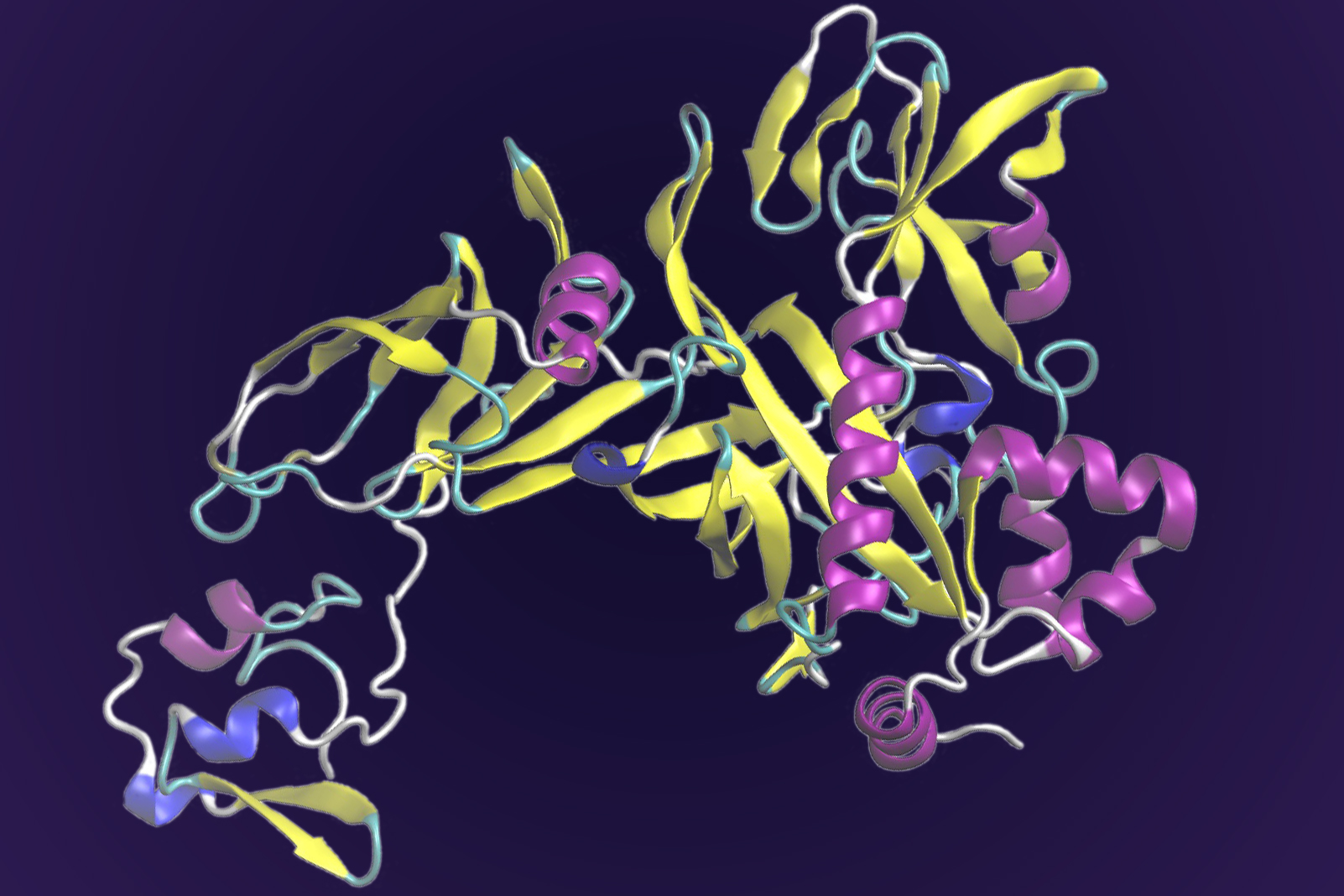
This protein drives key processes for sulfide use in many microorganisms that produce methane, including Thermosipho melanesiensis. Researchers used supercomputing and deep learning tools to predict its structure, which has eluded experimental methods such as crystallography. Image Credit: Ada Sedova, ORNL
The team also generated models of the 24,000 proteins at work in sphagnum moss. Sphagnum plays a critical role in storing vast amounts of carbon in peat bogs, which hold more carbon than all the world’s forests. These data can help scientists pinpoint which genes are most important in enhancing sphagnum’s ability to sequester carbon and withstand climate change.
Speeding scientific discovery
In search of the genes that enable sphagnum moss to tolerate rising temperatures, ORNL scientists start by comparing its DNA sequences to the model organism Arabidopsis, a thoroughly investigated plant species in the mustard family.
“Sphagnum moss is about 515 million years diverged from that model,” said Bryan Piatkowski, a biologist and ORNL Liane B. Russell Fellow. “Even for plants more closely related to Arabidopsis, we don’t have a lot of empirical evidence for how these proteins behave. There is only so much we can infer about function from comparing the nucleotide sequences with the model.”
Being able to see the structures of proteins adds another layer that can help scientists home in on the most promising gene candidates for experiments.
Piatkowski, for instance, has been studying moss populations from Maine to Florida with the aim of identifying differences in their genes that could be adaptive to climate. He has a long list of genes that might regulate heat tolerance. Some of the gene sequences are only different by one nucleotide, or in the language of the genetic code, by a single letter.
“These protein structures will help us look for whether these nucleotide changes cause changes to the protein function and if so, how? Do those protein changes end up helping plants survive in extreme temperatures?” Piatkowski said.
Looking for similarities in sequences to determine function is only part of the challenge. DNA sequences are translated into the amino acids that make up proteins. Through evolution, some of the sequences can mutate over time, replacing one amino acid with another that has similar properties. These changes do not always cause differences in function.
“You could have proteins with very different sequences—less than 20 percent sequence match—and get the same structure and possibly the same function,” Sedova said. “Computational tools that only compare sequences can fail to find two proteins with very similar structures.”
Until recently, scientists have not had tools that can reliably predict protein structure based on genetic sequences. Applying these new deep learning tools is a game changer.
Though protein structure and function will still need confirmation via physical experiments and methods such as X-ray crystallography, deep learning is shifting the paradigm by quickly narrowing the vast field of candidate genes to the most interesting few for further study.
Revolutionary tools
One of the tools in the deep learning pipeline is called Sequence Alignments from deep-Learning of Structural Alignments, or SAdLSA. Developed by collaborators Mu Gao and Jeffrey Skolnick at Georgia Tech, the computational tool is trained in a similar way as other deep learning models that predict protein structure. SAdLSA has the capability to compare sequences by implicitly understanding the protein structure, even if the sequences only share 10 percent similarity.
“SAdLSA can detect distantly related proteins that may or may not have the same function,” said Jerry Parks, ORNL computational chemist and group leader. “Combine that with AlphaFold, which provides a 3D structural model of the protein, and you can analyze the active site to determine which amino acids are doing the chemistry and how they contribute to the function.”
DeepMind’s tool, AlphaFold 2, demonstrated accuracy approaching that of X-ray crystallography in determining the structures of unknown proteins in the 2020 Critical Assessment of protein Structure Prediction, or CASP, competition. In this worldwide biennial experiment, organizers use unpublished protein structures that have been solved and validated to gauge the success of state-of-the-art software programs in predicting protein structure.
AlphaFold 2 is the first and only program to achieve this level of accuracy since CASP began in 1994. As a bonus, it can also predict protein-protein interactions. This is important as proteins rarely work in isolation.
“I’ve used AlphaFold to generate models of protein complexes, and it works phenomenally well,” Parks said. “It predicts not only the structure of the individual proteins but also how they interact with each other.”
With AlphaFold’s success, the European Bioinformatics Institute, or EBI, has partnered with them to model over 100 million proteins—starting with model organisms and those with applications for medicine and human health.
ORNL researchers and their collaborators are complementing EBI’s efforts by focusing on organisms that are critical to DOE missions. They are working to make the toolkit available to other users on Summit and to share the thousands of protein structures they’ve modeled as downloadable datasets to facilitate science.
“This is a technology that is difficult for many research groups to just spin up,” Sedova said. “We hope to make it more accessible now that we’ve formatted it for Summit.”
Using AlphaFold 2, with its many software modules and 1.5-terabyte database, requires significant amounts of memory and many powerful parallel processing units. Running it on Summit was a multistep process that required a team of experts at the Oak Ridge Leadership Computing Facility (OLCF), a DOE Office of Science user facility.
ORNL’s Ryan Prout, Subil Abraham, Nicholas Quentin Haas, Wael Elwasif, and Mark Coletti were critical to the implementation process, which relied in part on a unique capability called a Singularity container that was originally developed by Lawrence Berkeley National Laboratory. Mu Gao contributed by deconstructing DeepMind’s AlphaFold 2 workflow so it could make efficient use of the OLCF resources, including Summit and the Andes system.
The work will evolve as the tools change, including the advancement to exascale computing with the Frontier system being built at ORNL, expected to exceed a quintillion, or 1018, calculations per second. Sedova is excited about the possibilities.
“With these kinds of tools in our tool belt that are both structure-based and deep learning-based, this resource can help give us information about these proteins of unknown function—sequences that have no matches to other sequences in the entire repository of known proteins,” Sedova said. “This unlocks a lot of new knowledge and potential to address national priorities through bioengineering. For instance, there are potentially many enzymes with useful functions that have not yet been discovered.”
The research team includes ORNL’s Ada Sedova and Jerry Parks, Georgia Tech’s Jeffrey Skolnick and Mu Gao, and Jianlin Cheng from the University of Missouri, Columbia. Sedova virtually presented their work at the Machine Learning in HPC Environments workshop chaired by ORNL’s Seung-Hwan Lim as part of SC21, the International Conference for High Performance Computing, Networking, Storage and Analysis.
The project is supported through the Biological and Environmental Research program in DOE’s Office of Science and through an award from the DOE Office of Advanced Scientific Computing Research’s Leadership Computing Challenge. Piatkowski’s research on sphagnum moss is supported through ORNL’s Laboratory Directed Research and Development funds.—Kimberly Askey
Related Publication: Mu Gao et al., “High-Performance Deep Learning Toolbox for Genome-Scale Prediction of Protein Structure and Function.” Paper presented at the IEEE/ACM Workshop on Machine Learning in High Performance Computing Environments (MLHPC) Physics of Plasmas 28, no. 6 (2021), doi:10.1109/MLHPC54614.2021.00010.
UT-Battelle manages ORNL for the Department of Energy’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. The Office of Science is working to address some of the most pressing challenges of our time. For more information, please visit energy.gov/science.