Computational scientists at the Department of Energy’s Oak Ridge National Laboratory have published a study in the Journal of Chemical Theory and Computation that questions a long-accepted factor in simulating the molecular dynamics of water: the 2 femtosecond (one quadrillionth of a second) time step. The femtosecond is a timescale used by scientists to measure the ultrafast processes of atoms and molecules.
According to the team’s findings, using anything greater than a 0.5 femtosecond time step — the time interval at which a computer simulation is analyzed — can introduce errors in both the dynamics and thermodynamics when simulating water using a rigid-body description.
Because water is the most prevalent component of biomolecular simulations — from protein ensembles to nucleic acids — the team’s recommendation of a 0.5-femtosecond time step for better accuracy could cause some waves in the scientific community. The 2-femtosecond time step has been accepted as a standard in water simulations for almost 50 years.
“This has broad implications because water is the active constituent in cell biology. Water is the matrix of life, and all the simulations that we do on biological systems are always in water. But if you are simulating that fluid in a way that breaks a fundamental tenet of equilibrium statistical mechanics, that’s a problem,” said co-author Dilip Asthagiri, a senior computational biomedical scientist in ORNL’s Advanced Computing for Life Sciences and Engineering group.
Molecular simulations solve Newtonian equations of motion to elucidate how the molecules evolve over time. Of particular interest to researchers conducting such calculations is the determination of the resulting system temperatures. One of the tenets of statistical mechanics is that, if a system is in equilibrium, then the temperatures associated with its translational motion (movement along a line) and rotational motion should be the same. If those two temperatures differ, then the simulation is not in equilibrium. According to the team’s findings, that is the essential problem with using time steps longer than 0.5 femtoseconds to simulate water.
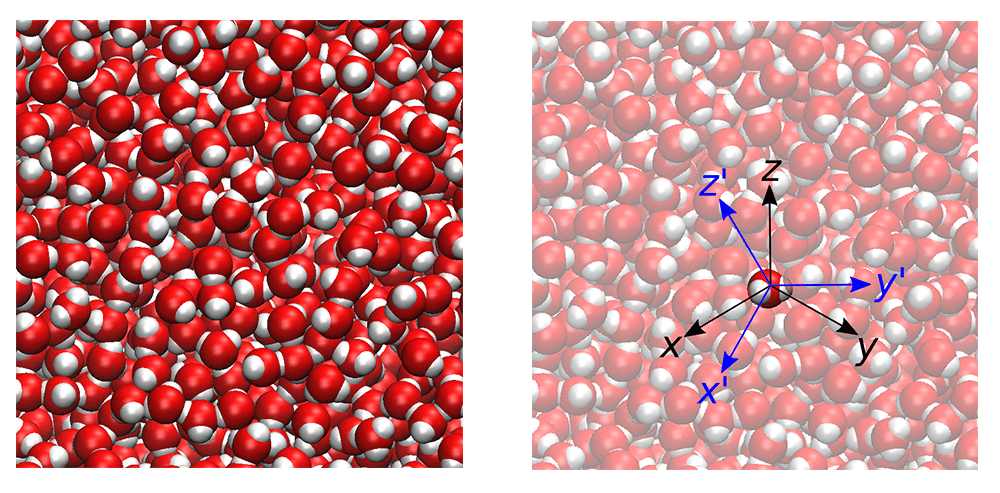
This schematic shows the rotational motion of a specific water molecule. At equilibrium, on average, the energy associated with rotational motion must equal the energy associated with the translation of the molecule as a whole. Credit: Dilip Asthagiri/ORNL, U.S. Dept. of Energy
The use of the 2-femtosecond time step in simulations arose from a paper published in 1977, when computing time was far more computationally expensive. Because the flexible bond between oxygen and hydrogen vibrates rapidly, the time steps necessary to accurately calculate that vibration are very small, requiring more computing time to capture enough intervals to study. Because that motion is the most rapid, that time step is the one that must be used in the evolution to obtain the right answer. The paper’s authors wanted to know, what if there was a way to use longer time steps and allow for fewer intervals and longer simulations? Those researchers proposed a rigid-body description of water to do just that.
“The 1977 work basically said that the vibrations of the oxygen-hydrogen bond can be decoupled from translation and rotation, and therefore freezing the vibrations by treating water as a rigid body should allow one to take a big time step,” Asthagiri said. “Since that time, the rigid-bond model has become the standard — the canonical way that scientists look at this.”
But Asthagiri discovered that using this method can cause discrepancies in the temperatures between the water molecules’ translational and rotational motions, meaning the simulation may be producing incorrect results.
“What Dilip found is that going with too long of a time step, you tend to get inaccurate values for both the thermodynamics and the dynamics of the motion of water, which is the medium in which all these molecules move. In effect, you can get a false friction, either too large or too small, due to this approximation of too long of a time step. And if you have the friction off, that means the motion of these molecules is going be off, too,” said co-author Tom Beck, section head of Science Engagement in the National Center for Computational Sciences at ORNL.
Asthagiri first noted this disparity in temperatures as a research professor at Rice University in 2021. He and a graduate student were simulating water in the supercooled regime and found that the average temperature in the log file was lower than the setpoint temperature.
“It was a 1 Kelvin difference, and you can easily ignore it, but it was systematically seen at different temperatures. And that was the clue that there was something off — okay, maybe one temperature, but multiple temperatures with the same behavior? There must be something wrong,” Asthagiri said.
After joining ORNL in 2022, Asthagiri began examining rotation and translation separately rather than using the site coordinates and velocities, which are standard quantities that biomolecular simulation codes produce. Incidentally, formulating the equations of those motions separately was the approach used by the authors of the very first paper ever written on simulating water in 1971. Those authors recommended a time step of 0.4 femtoseconds.
“We need to go back to the original work in terms of being careful. There is nothing wrong with doing site velocities, but if you do it as site velocities, then you need to take a time step that is small enough that temperatures between translation and rotation are the same, on average,” Asthagiri said.
Computational scientists can easily make the change to 0.5-femtosecond time steps, should they choose to do so, although it would also result in shorter simulations because of longer computing times.
“It’s just one flag in the input script — 2 to 0.5. It’s a very simple switch, but now the problem is you have to use more computing time, that’s all. But the computing power is available now,” Asthagiri said.
Asthagiri has presented the study’s findings to colleagues at the Telluride Science & Innovation Center and the online Statistical Thermodynamics & Molecular Simulations Seminar Series.
“When I presented the work to an online statistical thermodynamics seminar series, the first reaction was a little bit of a shock. It’s going to take time to sink in,” Asthagiri said.
Asthagiri will present the results at another workshop co-organized by Beck for the Centre Européen de Calcul Atomique et Moléculaire on May 6-8 in Pisa, Italy.
Related Publication:
D. Asthagiri and T. Beck, “MD Simulation of Water Using a Rigid Body Description Requires a Small Time Step to Ensure Equipartition,” Journal of Chemical Theory and Computation (2024): https://doi.org/10.1021/acs.jctc.3c01153.
UT-Battelle manages ORNL for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time. For more information, visit energy.gov/science.