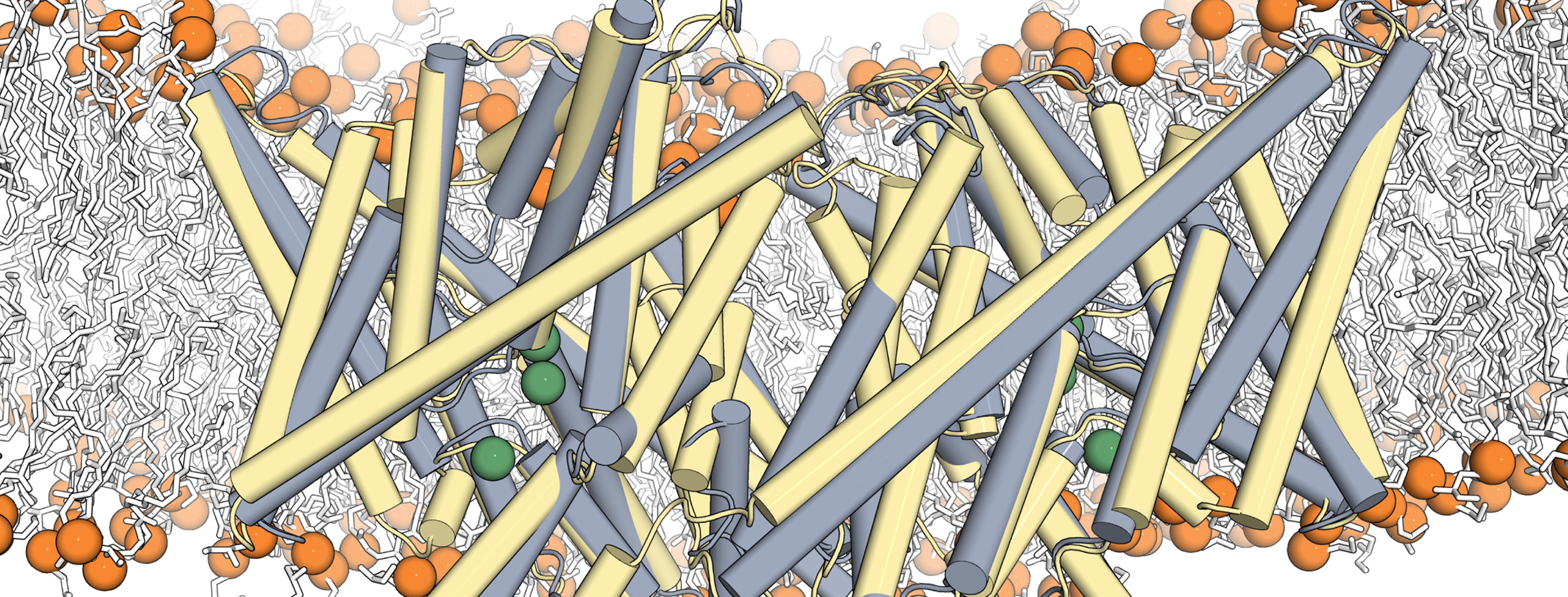
A team of researchers from Weill Cornell Medical School’s Department of Physiology and Biophysics has developed a new computational technique to conduct molecular dynamics simulations of proteins at the specific concentration of hydrogen ions (pH) at which they function. Called the Equilibrium Constant pH (ECpH) method, it presents a significant advance in the ability of computational simulations to more accurately represent the way in which a human cell’s membrane proteins look and how they function under different conditions encountered in the life of the cell.
Harel Weinstein, director of the Institute for Computational Biomedicine at Weill Cornell Medicine, along with co-authors Ekaterina Kots (who developed the method) and Derek M. Shore, revealed the details of ECpH and expounded on the results of its application in a paper recently published in the journal Molecules. The team used the Summit supercomputer at the Oak Ridge Leadership Computing Facility (OLCF), a US Department of Energy (DOE) Office of Science User Facility at Oak Ridge National Laboratory (ORNL), to test the performance of its ECpH code and carry out extensive production simulations.
“That many proteins function at a variety of pH levels in different ways is a problem that has embittered the lives of biologists for a very long time. Anybody interested in understanding protein structure/function relations under various pH conditions couldn’t do anything about it because neither structure determination nor computation were able to help them in a significant fashion,” Weinstein said. “Now, what we have developed is a novel method to carry out molecular dynamic simulations in a manner that allows us to look at the protein at the entire range of pH and predict pH-induced conformational changes.”
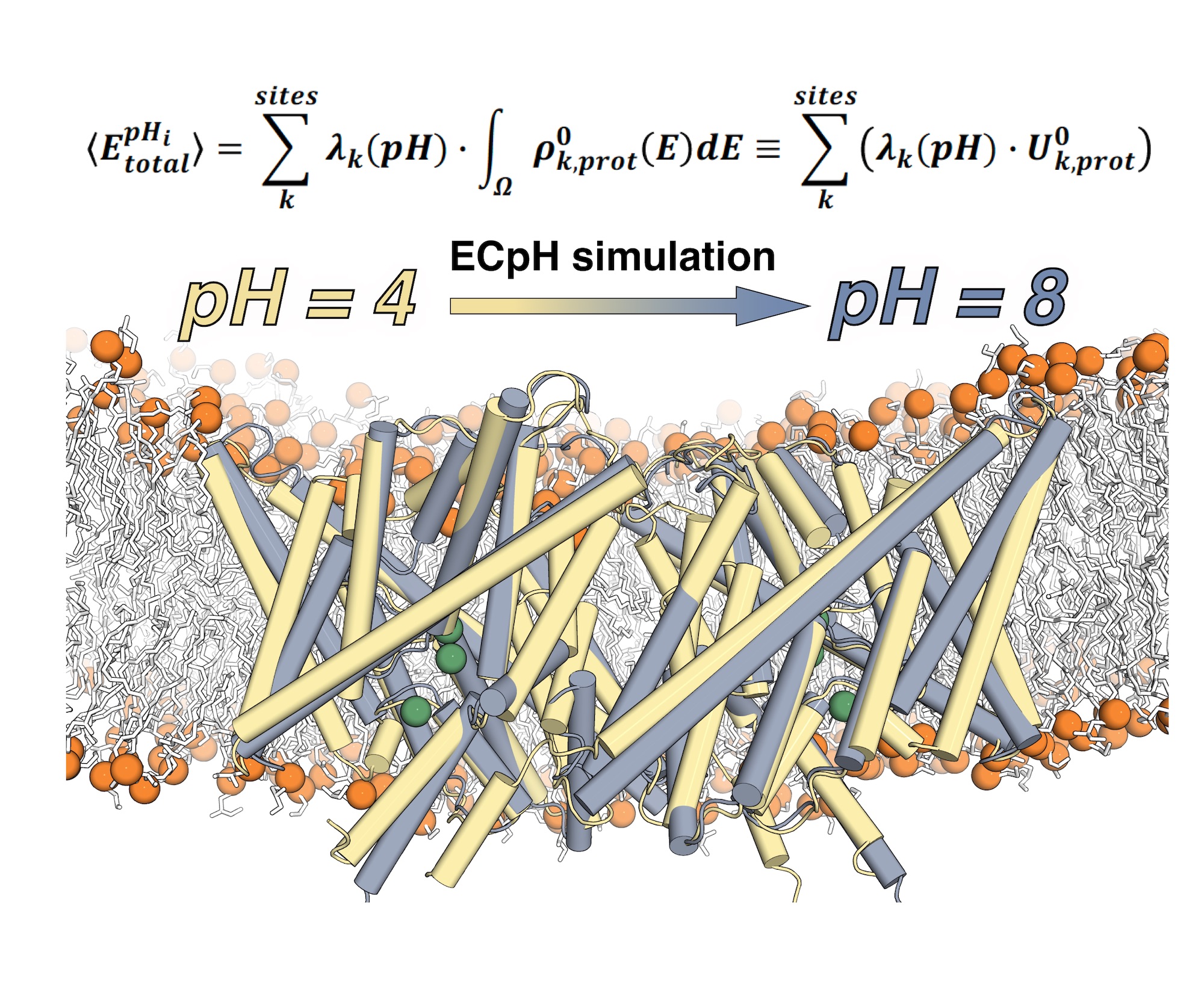
This representation shows the ECpH simulated conformational transition of a CLC1-ec molecular dimer embedded in a membrane, from its conformation at pH4 (in yellow) to the one it adopts at pH8 (in grey). The average value of the total energy of a system with multiple pH-dependent protonation sites (k) and protonation probabilities contained in the set λ(pH) is calculated from the ECpH simulation trajectory as shown. Image courtesy of Ekaterina Kots, Weill Cornell Medical School.
Proteins are the workhorses of our body’s cells, performing vital functions we can’t live without—everything from helping form antibodies to transporting nutrients. The individual role of each protein can be determined by studying its unique structure. Proteins that sit in the external membranes of cells are tasked with recognizing changes outside of the cell, such as the introduction of a drug, and communicating them inward so the cell can respond.
These proteins can stay lodged in cell membranes because their external shell is formed by hydrophobic amino acids. Interspersed among these hydrophobic components of the protein are positive and negative charges that play a major role in recognition of outside signals and in the rearrangement of the molecule that allows it to transmit information into the cell. However, these charged residues are susceptible to changes in pH levels.
Different pH levels—a measure of acidity in fluids—in and around the cells trigger different actions by proteins. If the pH changes, this can cause some of the residues that are negatively charged to become neutral by picking up a proton. This change in charge distribution will cause the structure of the protein to change as well. This conformational change is almost invariably essential for function.
“In fact, nature is using the conformational change that occurs at a changed pH to activate or deactivate molecules, and this happens in both normal function of the proteins and in disease. Controlling pH is a major way nature has devised to control the activity of its proteins,” Weinstein said. “Now, what is the mechanism of activation by pH changes? This was hard to establish. But we can now because thanks to the computational power of modern leadership-class resources: it unfreezes the protein entirely. We can say what happens to the entire protein under the new conditions and what the mechanism is by following it from the beginning to the end.”
Weinstein and his team also ran ECpH on Summit to verify the results of another new computational technique developed by Weill Cornell Medicine scientists for high-resolution analysis of biomolecules: a new method of measurement with atomic-force microscopy called localization atomic force microscopy or LAFM. Current state-of-the-art techniques to examine the dynamic behavior of biomolecules face limitations owing to their static approaches: cryo-electron microscopy freezes the molecules for study, whereas x-ray crystallography relies on being able to crystallize the molecules, which is a complicated procedure for most biomolecules. LAFM, on the other hand, is a direct, room-temperature visualization of membrane molecule dynamics.
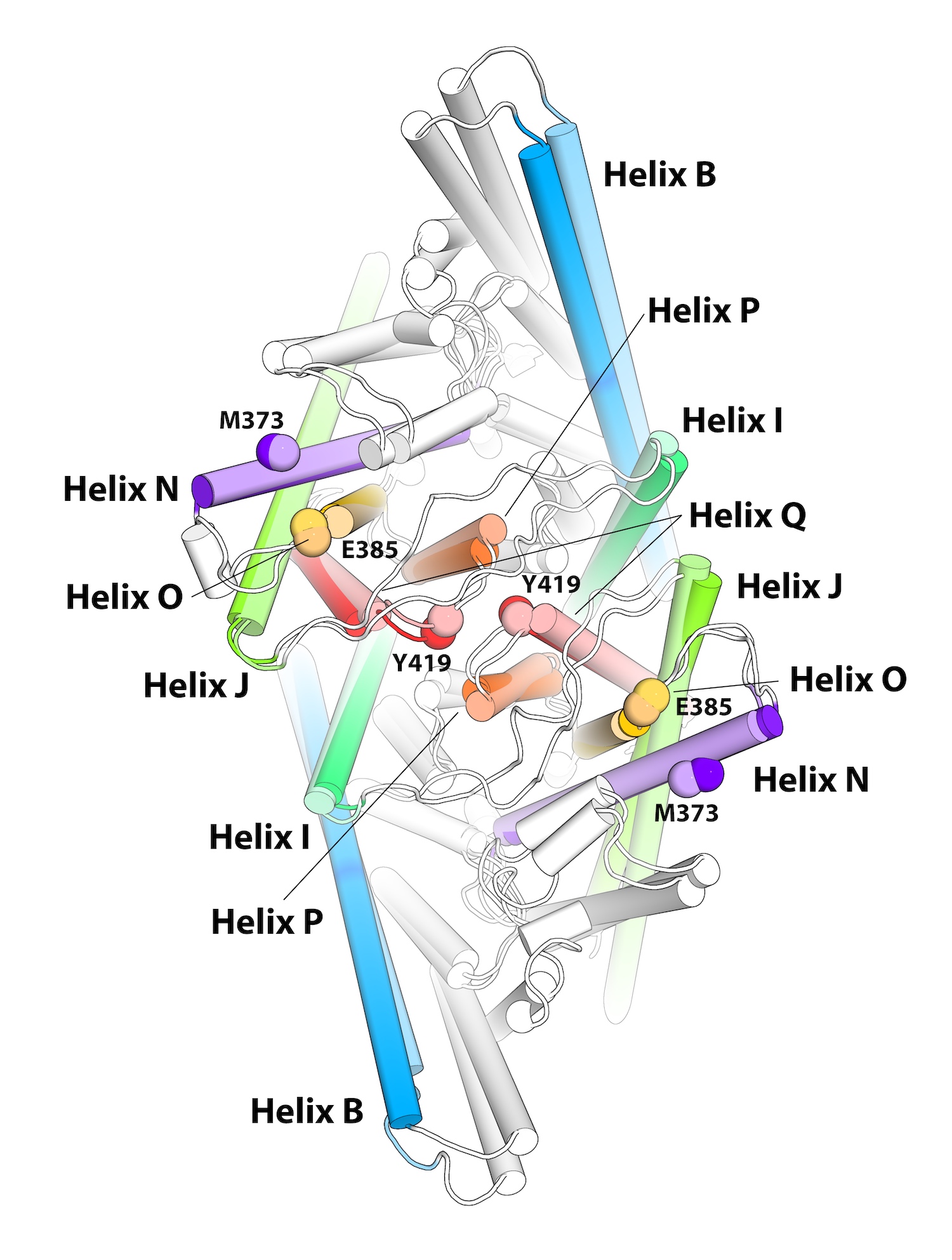
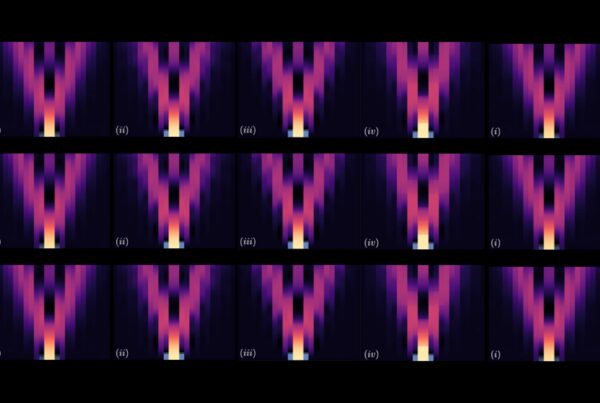
The specific rearrangements in the structure of the CLC1-ec dimer revealed by the ECpH simulation served to interpret and explain the spatial reorganization observed with the new method measuring the dynamics of membrane proteins with atomic force microscopy: LAFM, or localization atomic force microscopy. (The component helices are shown as identified colored cylinders that change in position and orientation.) Image courtesy of Ekaterina Kots, Weill Cornell Medical School.
“So, what we now have is a technique that—in real time, without freezing—allows us to look at the molecule as it behaves in the membrane. That is an enormous advance,” Weinstein said. “Like every other methodology, it has its limitations. The limitation is that this technique is based on atomic force microscopy and for membrane proteins it reports only changes that are detectable above the membrane surface.”
Although standard atomic force microscopy can sketch out details of molecules in situ using an atom-sized “needle,” it is limited to proteins that protrude from a cell’s membrane surface. And because those proteins are moving, AFM images tend to be blurry. To counter that effect, LAFM applies localization algorithms to the spatial fluctuations of features to create high-resolution images. Being able to obtain the predicted changes in structure at various pH values from ECpH simulations helped interpret LAFM results in the Nature article.
Together, these innovations produce structural and dynamic information that exceeds the image resolutions of current physiological and computational methods of analysis. They promise to bring a new understanding of molecular mechanisms.
Support for this project came from DOE’s Innovative and Novel Computational Impact on Theory and Experiment program with an allocation (BIP109) of compute time at the OLCF, which is a DOE Office of Science User Facility supported under Contract DE-AC0500OR22725.
Related Publications:
Kots, E., D. M. Shore, H. Weinstein. “An Equilibrium Constant pH Molecular Dynamics Method for Accurate Prediction of pH-Dependence in Protein Systems: Theory and Application.” Molecules, November 18, 2021.
Heath, G. R., E. Kots, J. L. Robertson, S. Lansky, G. Khelashvili, H. Weinstein and S. Scheuring. “Localization Atomic Force Microscopy.” Nature, June 16, 2021.
UT-Battelle LLC manages ORNL for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time. For more information, visit https://energy.gov/science.