Can something as simple as shape fully determine whether or not proteins will bind together? Scientists are commissioning supercomputers to find out.
A team led by Sharon Glotzer, distinguished professor and department chair of chemical engineering at the University of Michigan (UM), used the 200-petaflop Summit supercomputer at the US Department of Energy’s (DOE’s) Oak Ridge National Laboratory (ORNL) to model lock-and-key interactions between proteins to study their binding behaviors. The results, published in Soft Matter, revealed that some proteins do, in fact, bind based on shape alone.
“We’ve demonstrated that something as simple as shape is able to predict protein interactions that are sometimes really complex,” said Jens Glaser, computational scientist in the Advanced Computing for Chemistry and Materials group at the Oak Ridge Leadership Computing Facility (OLCF). “This first demonstration has led us to believe that shape has been an unappreciated ingredient in many protein assembly processes.”
The results could have numerous applications in biological research. For example, the approach might be used to screen drugs for disease or provide scientists with information about how to use proteins as building blocks to design new biological materials.
“This exciting study demonstrates the power of shape complementarity in the prediction of protein-protein interfaces,” said Dr. Stephanie McElhinny, program manager at the US Army Combat Capabilities Development Command’s Army Research Laboratory, referring to the favorable spatial relationship between two compatibly shaped proteins. “Computational models that accurately predict these interfaces will support the future design of advanced protein-based materials with active and responsive properties, such as light-harvesting protein-based plastics that could function like an artificial leaf for power generation.”
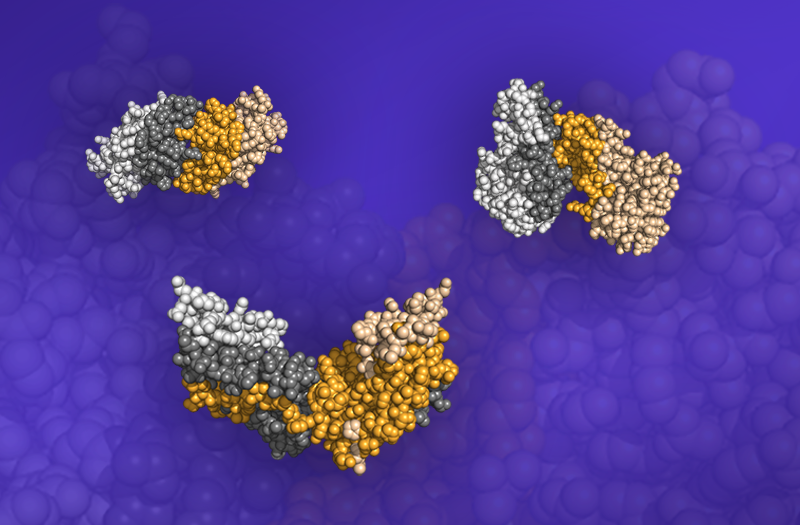
Three dimers, protein structures consisting of two bound proteins, from the Dockground database. The interfaces at which the proteins meet are shown as the darkened regions. Image credit: ORNL
Supercomputers reveal shape is key in some proteins
For proteins to successfully bind to one another, one of them acts as a ligand, a molecule that attaches to a target protein, and one of them acts as a receptor, the molecule that receives the ligand. This process involves complex chemical interactions, in which molecules share bonds and change their configurations upon binding.
Glotzer’s team wanted to see whether they could predict this molecular binding based on shape alone, ignoring the interactions between proteins. From a database of more than 6,000 protein pairs, the team tested 46 pairs that are known to bind to one another and simulated their assembly on Summit. The team performed the simulations under the INCITE (Innovative and Novel Computational Impact on Theory and Experiment) program.
Like multiple tennis balls being thrown at a single target, the simulations modeled multiple ligands being tossed at a single, fixed target receptor. Out of the 46 pairs tested, they found 6 pairs that performed well—more than 50 percent of the time they successfully assembled based purely on their complementary shapes.
“We looked at the interfaces where the proteins bound together to see how similar they were to their real-life interfaces, and then we determined the cutoff to see how many pairs were good predictors of the real interfaces,” said Fengyi Gao, PhD candidate at UM. “We found that 13 percent of these protein pairs could bind based on shape alone.”
The team then built a machine-learning model that could determine which proteins are able to assemble solely based on shape. Combining their initial model with such machine-learning tools will help them understand what information is needed for protein pairs that cannot assemble based on shape complementarity alone.
Running proteins in parallel
To model multiple reversible binding processes of 46 protein pairs under different parameters, they needed two days of computational time and more than 3,000 GPUs—an amount that only a supercomputer like the OLCF’s Summit could provide. The OLCF is a DOE Office of Science User Facility at ORNL.
As part of the HOOMD-blue computational code that was used to run the simulations, Glaser, who was previously an assistant research scientist in Glotzer’s group at UM, developed an algorithm that simulated the proteins in the presence of many small particles. But Glaser found a way to model only the motion of the proteins the team was interested in, avoiding unnecessary and expensive calculations for the solvent molecules around them.
“I ran the code in parallel so that many different parameters, iterations of the same system, and different proteins could be distributed across the GPUs,” Glaser said. “This allowed us to easily make use of Summit’s parallel computing capabilities.”
Using Summit, the team captured six protein pairs that bound based only on shape complementarity, with one of them achieving binding more than 94 percent of the time.
A simulation trajectory of a pair of proteins. A team led by Sharon Glotzer at the University of Michigan simulated a receptor protein (red) fixed in a simulation box and 50 independent binding processes. The ligand proteins are colored by how closely their binding pose is to the native configuration, with yellow being far from the binding pose and purple being the best prediction of the binding pose. At the end, only one protein out of 50 misses the correct configuration, revealing a 98% yield. Video Credit: Fengyi Gao, University of Michigan
“It was quite surprising to us that such a simplified model could correctly select just that one pose that they assume out of the many hundreds or more poses that compete,” Glaser said. “We were expecting that much more would be necessary to reproduce the real binding pose for these protein pairs.”
Models may aid in drug screening
The team plans to study more proteins that can also bind based on shape—or form even higher order structures. The team’s current study explored only protein dimers, which consist of two proteins bound together, but the team wants to know the limitation for how protein shapes can evolve to form hierarchical protein structures.
“Before we did this study, I actually didn’t expect proteins could form dimers based on shape alone,” said Fengyi Gao, PhD candidate at UM. “But now, we’ve found that this works, and we can study more complex structures or even combine this with other approaches, like machine learning, to see which features we need to enable the correct binding.”
The team hopes they can eventually predict the binding of protein-protein interfaces in protein clusters or protein crystallization structures.
“We think we can adapt this approach to something like drug screening in the future,” Gao said. “In addition to that, we hope that this shape-based model can serve as a basis of studying protein assembly in general.”
Sharon C. Glotzer is the John W. Cahn Distinguished University Professor of Engineering, the Anthony C. Lembke Department Chair of Chemical Engineering, and the Stuart W. Churchill Collegiate Professor of Chemical Engineering at the University of Michigan. She is also Professor of Materials Science and Engineering, Physics, Applied Physics, and Macromolecular Science and Engineering.
This work was supported by the US Army Research Laboratory and the US Army Research Office.
Related Publication: Fengyi Gao, Jens Glaser, and Sharon Glotzer, “The Role of Complementary Shape in Protein Dimerization,” Soft Matter (2021), doi:10.1039/D1SM00468A.
The research was supported by DOE’s Office of Science. UT-Battelle LLC manages Oak Ridge National Laboratory for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time. For more information, visit https://energy.gov/science.