Francisella tularensis, a bacterium that causes the illness tularemia (also known as “rabbit fever”), is one of the most hostile organisms on the planet. Once the bacterium enters the body and tularemia sets in, an infected person can experience a range of symptoms, from ulcers to tonsillitis to pneumonia that can turn fatal.
The bacterium is spread through contact with infected animals, drinking contaminated water, tick and deer fly bites, and inhalation in certain agricultural environments. It also has the potential to be a serious bioterrorism threat; it can be easily spread via aerosol, and as few as 10 organisms can cause the disease. Despite its virulence, scientists have not identified the best target in Francisella’s genetic material for a vaccine. Understanding the protein structures that are encoded by the bacterium’s genetic material is crucial for developing vaccines and treatments against these structures, but getting a detailed look at them has been challenging because the bacterium is difficult to isolate.
Now, a team of scientists led by Abhishek Singharoy, assistant professor at Arizona State University (ASU), has used the IBM AC922 Summit supercomputer at the Oak Ridge Leadership Computing Facility (OLCF) to simulate the structure of a possible drug target, a lipoprotein (or fat-transporting protein) called Francisella lipoprotein 3 (Flpp3), to reveal the different possible states the protein can exist in. The simulations were guided by experiments that employed different methods to understand the protein’s structure.
“When a drug discovery pipeline is created, you need information for the druggability of multiple conformations, or states,” Singharoy said. “You might only find a handful of conformations that are druggable.”
The team has since begun virtually screening compounds that might easily interfere with the Flpp3 protein and prevent Francisella from spreading. Identifying drugs that could bind to Flpp3 is the next step towards preparation for biological attacks.
Summit settles a mystery
Among the methods scientists can use to determine the structural states of molecules are nuclear magnetic resonance (NMR) and x-ray crystallography (XRC). NMR uses a varying magnetic field to calculate the distance between atoms and identify compounds, and XRC uses x-ray diffraction images from crystals, formed by packing together molecules, to determine the molecular structure.
Scientists previously identified Flpp3 as a protein that can determine the virulence of tularemia, and in 2015 Petra Fromme, director of the Biodesign Center for Applied Structural Discovery at ASU, and ASU graduate student James Zook used NMR to capture its structure. Then, to get an even better look at Flpp3’s structure, they took the protein to the Linac Coherent Light Source (LCLS), a US Department of Energy (DOE) Office of Science User Facility at the SLAC National Accelerator Laboratory. The team performed XRC on the protein using the LCLS, and results showed a difference in the NMR and XRC structures—one showed the open protein and the other showed the closed protein. But what was happening in the middle?
“In the body, proteins spontaneously open and close, but in an experiment, you only get one or the other,” said Josh Vermaas, a computational scientist in the Scientific Computing Group at the OLCF. “Identifying states along the process between those two end states allows scientists to target the in between states as well or prevent it from opening or closing.”
After earning computing time under the Innovative Novel and Computational Impact on Theory and Experiment program, the team gained access to Summit, a 200-petaflop system at the OLCF, a DOE Office of Science User Facility located at DOE’s Oak Ridge National Laboratory. Using a computational method called molecular dynamics—which calculates the movement of molecules in time and space—Singharoy, with ASU postdoctoral researchers Mrinal Shekhar and Chitrak Gupta, modeled the mystery states between the XRC and NMR results. After performing thousands of simulations consisting of more than 100,000 atoms each, the team found that the protein has a cavity that a drug molecule could take advantage of and fill while the protein is busy opening or closing.
“Sometimes, drugs have to have the right shape to fit the pocket that’s formed by the protein,” Vermaas said. “A lot of drugs that bind to membrane proteins will activate a protein or turn it off or impact it one way or another.”
Discovery in silico
Armed with an exhaustive set of models for Flpp3, the team screened drug compounds using the ZINC database, a collection of commercially available compounds. After finding one possible molecule and then modifying it to increase possible bonding sites, the team devised a drug compound that specifically inserts itself deep into the protein cavity of Flpp3.
“We found that the different states of this protein can be overcome at room temperature, meaning this is something that can easily be studied in a laboratory setting,” Singharoy said.
“The next step is to investigate this as not only a binding partner to the protein but also as an agent against tularemia.”
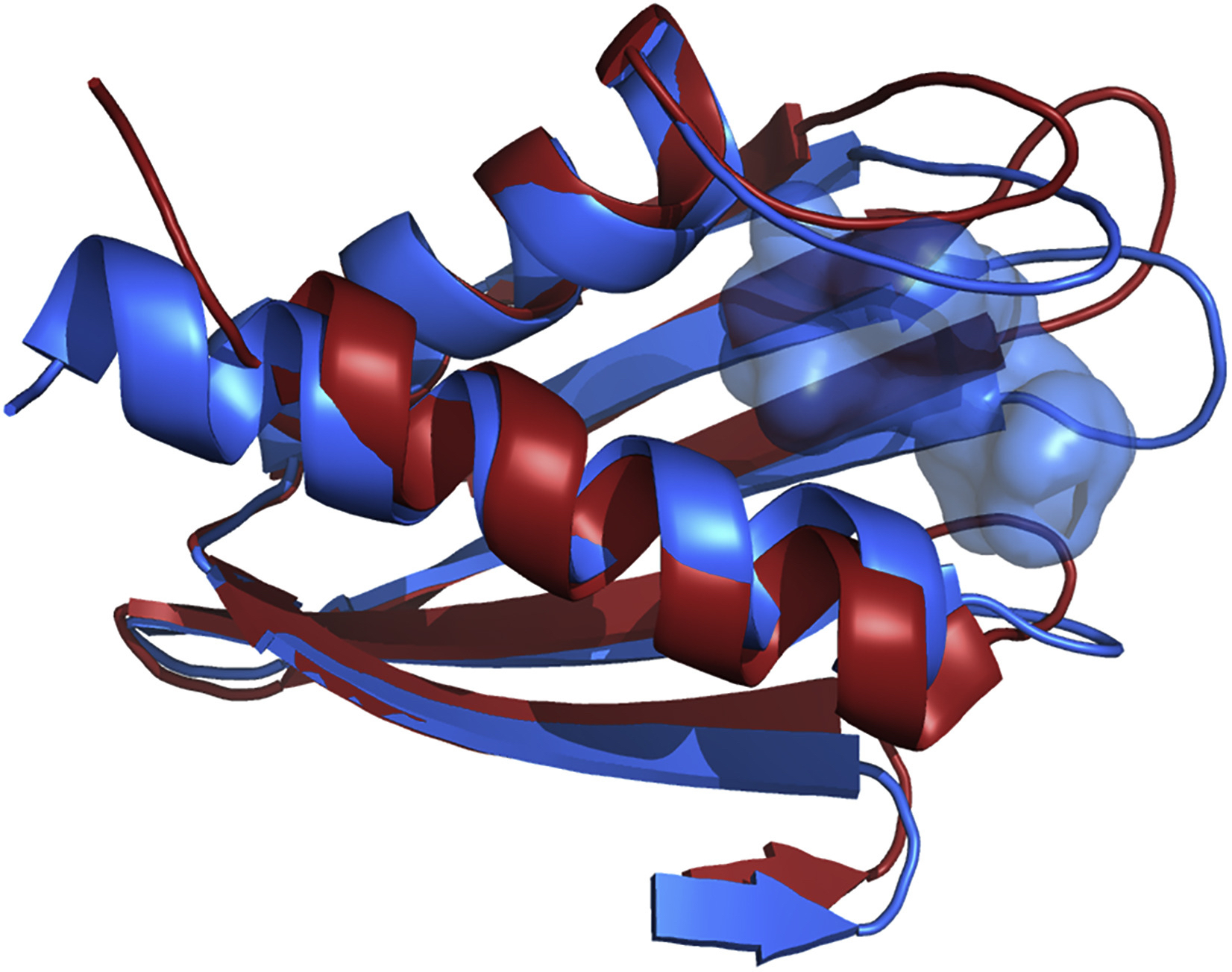
Flpp3 structures from NMR (red) and XRC (blue), from 2MU4 and 6PNY in the Protein Data Bank, respectively. There is no cavity observed in the XRC structure, which would appear as a red, semi-transparent surface, but a cavity is shown in the NMR structure (blue, semi-transparent surface). Image Credit: Abhishek Singharoy, ASU
The team hopes that the new understanding of the protein’s different conformations, combined with the virtual screening, will lead to antibiotics that can target tularemia. Future studies will look at the antimicrobial effects of the drug compounds that bind to Flppp3.
“Ultimately, these results are ultimately allowing us to look at drug targets in a more realistic way than ever before,” Singharoy said.
Related Publication: Zook, J. et al. “XFEL and NMR Structures of Francisella Lipoprotein Reveal Conformational Space of Drug Target against Tularemia.” Structure 28 (2020): 1–8. doi:10.1016/j.str.2020.02.005.
UT Battelle LLC manages ORNL for DOE’s Office of Science. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, please visit https://energy.gov/science.