A map of gene expression correlation triangles, with positive correlations (blue edges) between Kalanchoë genes (dark green nodes) and pineapple genes (yellow nodes) and negative correlations (red edges) between Kalanchoë or pineapple genes and Arabidopsis genes (light green nodes).
During normal photosynthesis, plants capture sunlight and carry out respiration during the day, taking in carbon dioxide that will be used to form carbohydrates. But a small percentage of plant species keep their stomata closed during the day and open them to capture carbon dioxide at night to conserve water in a form of photosynthesis called crassulacean acid metabolism (CAM). Scientists are interested in studying CAM for its bioengineering applications, but insights into this unique photosynthetic process lie locked inside the genomes of these water-efficient plants.
A team led by computational biologist Daniel Jacobson at the US Department of Energy’s (DOE’s) Oak Ridge National Laboratory (ORNL) has developed a new method of comparing genes of CAM plants with genes of “normal” plants—called C3 plants—that will provide scientists with a detailed understanding of the mechanisms behind CAM. The team is part of a group of computational and experimental scientists led by ORNL’s Xiaohan Yang across the biological and chemical science domains who have a long-term goal of engineering CAM into bioenergy and food crops to render them tolerant to drought.
“We are trying to find the evolutionary patterns across how CAM has evolved in different species,” Jacobson said. CAM plants exhibit convergent evolution, meaning unrelated species of CAM plants have assumed similar traits over time to adapt to environments in which water is scarce.
The team compared two CAM plants—pineapple and a tropical succulent called Kalanchoë—with a C3 plant called Arabidopsis thaliana. A small flowering plant, Arabidopsis is well understood in the biological community, serving as the default model for many plant genomics studies.
The team used resources at the Oak Ridge Leadership Computing Facility (OLCF), a DOE Office of Science User Facility at ORNL, to compare billions of genes from the three plants. Comparing two CAM species with a non-CAM species allowed the team to successfully find correlation patterns among genes, bringing scientists one step closer to leveraging CAM mechanisms for feedstocks.
Searching for “triangles”
Not all species possess the same genes, so biologists often group genes into families to study and compare their functions and relations across different species. Gene families consist of genes with similar sequences of nucleotides—the building blocks of DNA and RNA—and they typically have the same or similar functions in the cell. Scientists can identify the expression patterns in different species by comparing genes from the same family that are regarded as equivalent.
Jacobson and his team specifically focused on time-related patterns of gene expression as part of a larger CAM project at ORNL that aimed to identify common sets of genes in CAM plants. Jacobson’s team compared plant data that described the abundance of expressed (activated) genes throughout the day based on sequenced RNA molecules in each plant’s tissue. The researchers studied expressions of similar genes at different times of day for each of the three plants to determine whether gene “enrichment,” or abundance, occurred in the daytime or nighttime.
“Comparing the expressions of the genes in CAM versus C3 plants at daytime versus nighttime is important because the magic of CAM is that these plants are fixing carbon at night,” Jacobson said, describing the first step in the process plants use to turn carbon dioxide into food molecules such as glucose. “That’s why plants like Kalanchoë, pineapple, and cacti are really water-efficient. They’re doing this at night.”
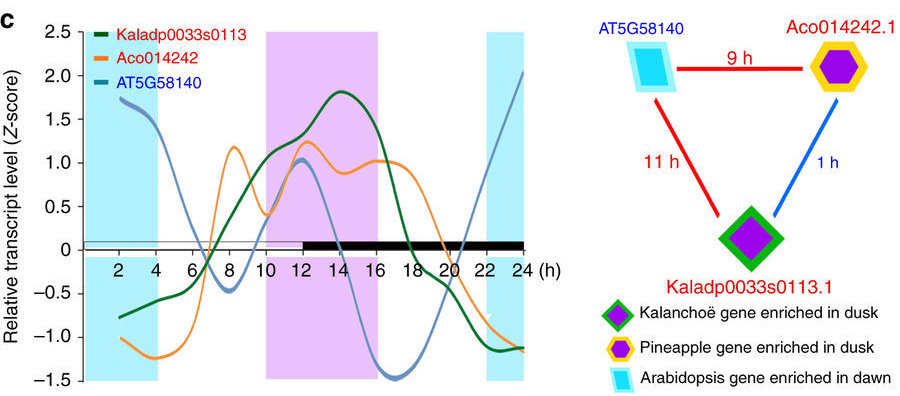
Comparison of transcript expression patterns in Kalanchoë, pineapple, and Arabidopsis genes. (left) Expression profiles. (right) Enrichment triangle network, showing the Kalanchoë and pineapple genes had significantly enriched expression in the same time-window, and the Arabidopsis gene had significantly enriched expression in the opposite time-window. The numbers of hours represent the time shifts in the expression pattern between the genes connected by each edge.
The group ran comparisons of the genes on OLCF resources under a Director’s Discretionary allocation, using a method based on the Fisher exact test, which can determine to what degree two variables are statistically related. The team searched for gene families in which (1) the Arabidopsis gene had an expression pattern opposite that of the genes of the pineapple and Kalanchoë, and (2) the pineapple and Kalanchoë shared the same expression patterns. The result was a collection of gene “triangles”—and a new method of studying time-related patterns of gene expression across species.
“The Fisher enrichment triangle method is a novel way of looking at this data across species,” Jacobson said. “When we look at opposite expression patterns, we can see what genes might be functionally related and look at which genes probably had a common ancestor. This method captures that in a way that other methods simply wouldn’t be able to, due to noise in the expression.”
Big genomes, big data, big resources
Comparing the genes of three plants may not sound like much to the average person, but Jacobson’s team knew the project would require a high-performance computing (HPC) system. Performing “all-versus-all” comparisons for roughly 30,000–40,000 genes of each plant—comparing each plant’s data with the others—necessitated supercomputing. Jacobson and his team used OLCF resources to refine their comparison method, starting with individual genes and then constraining them into their respective gene families.
The team initially performed more than 20 billion comparisons using mcxarray, a part of the MCL-Edge software package, to parallelize the code for OLCF resources. Mcxarray can compare rows and columns of data, ignoring positions where either one has missing data.
The team ultimately condensed the problem into gene families and compared similar genes using a code written by team member Deborah Weighill, a graduate student at the Bredesen Center for Interdisciplinary Research and Graduate Education, a collaboration between ORNL and the University of Tennessee. Weighill wrote the code in the Perl programming language, often used in bioinformatics. In addition to the OLCF, the team also used ORNL’s Compute and Data Environment for Science, an infrastructure including data resources and visualization tools for HPC, to analyze and store its large datasets.
The team’s results enabled scientists at ORNL to identify 54 genes that showed convergent evolution in CAM species, providing insight into CAM mechanisms that may prove useful for bioengineering agronomic plants that are more water-efficient.
“The long-term goal is giving bioenergy feedstock crops the ability to live in these less-favorable environments,” Jacobson said. “But this same concept can be applied to any other plant. The agronomic impact of that sort of engineering could be quite significant over time by making currently unavailable agricultural land suddenly available.”
The Fisher triangle enrichment method could be applied to study any mechanism across species, according to team members, who believe the method may even have potential applications in human clinical genomics studies, such as those related to diseased versus nondiseased populations.
“On the one hand, we are finding out how different plants work on a cellular level, and that’s extremely exciting. But it also has a broader impact,” Weighill said. “Whether this has an agronomic application or a clinical application for human studies, we’re excited about both.”
The research was supported by DOE’s Office of Science, the UK Biotechnology and Biological Sciences Research Council, and ORNL’s Laboratory Directed Research and Development Program. Genomic sequencing was performed at the DOE Joint Genome Institute, a DOE Office of Science user facility at DOE’s Lawrence Berkeley National Laboratory.
Related Publication: Xiaohan Yang, et al., “The Kalanchoë Genome Provides Insights into Convergent Evolution and Building Blocks of Crassulacean Acid Metabolism,” Nature Communications 8 (2017): 1899, doi:10.1038/s41467-017-01491-7.
ORNL is managed by UT-Battelle for the Department of Energy’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time. For more information, please visit https://science.energy.gov/.