
A team of computational scientists at the Department of Energy’s Oak Ridge National Laboratory has generated and released datasets of unprecedented scale that provide the ultraviolet visible spectral properties of over 10 million organic molecules. Understanding how a molecule interacts with light is essential to uncovering its electronic and optical properties, which in turn have potential photoactive applications in products such as solar cells or medical imaging systems.
Using high-performance computing resources at the Oak Ridge Leadership Computing Facility, the ORNL team ran quantum chemistry calculations to generate the vast datasets. For each of these organic molecules, the team ran atomistic material modeling calculations with various approximations to compute different excited-state properties of interest. The team’s findings were published in Nature Scientific Data.
The ultimate intended use for the open-source datasets is to train a deep learning model to identify molecules with tailored optoelectronic and photoreactivity properties, an approach that is much faster and easier to conduct than current methods.
“The use of DL models for molecular design is essential because the chemical space that must be explored for the search of these molecules is extremely large,” said lead author Massimiliano Lupo Pasini, a data scientist in ORNL’s Computational Sciences and Engineering Division.
“Both experiments and existing first-principles calculations, which are based on the physical laws that determine how matter and energy interact at the subatomic level, are simply unaffordable for different reasons. Experiments are labor intensive, and first-principles calculations can easily slam supercomputing facilities. But DL models provide very promising tools to overcome these barriers,” Lupo Pasini said.
The project got off the ground when Stephan Irle, leader of ORNL’s Computational Chemistry and Nanomaterials Sciences group, identified the ultraviolet-visible spectrums of molecules as a useful property to predict with DL models. Building a DL model sufficiently complex to identify desirable molecular properties requires training it with huge volumes of data that explore all different regions of chemical space. The more data collected, the more the DL model trained on it can achieve the necessary robustness and generalizability to function effectively. However, collecting such large volumes of scientific data for scalable DL may present data-flow issues, especially at facilities with multiple users like the OLCF, a DOE Office of Science user facility located at ORNL.
“One challenge that occurs when generating large volumes of data is that the number of files to manage increases drastically. If not managed correctly, such a large volume of data can compromise the functioning of the parallel file system, which is an important component of state-of-the-art HPC facilities,” Lupo Pasini said.
To address this challenge, Lupo Pasini collaborated with ORNL computer scientist Kshitij Mehta to develop a scalable workflow software that ensures that the files generated by the quantum mechanics code are properly handled without stressing the file system, such as the OLCF’s Orion, which is a shared resource that handles the input, output and storage of data on supercomputer systems.
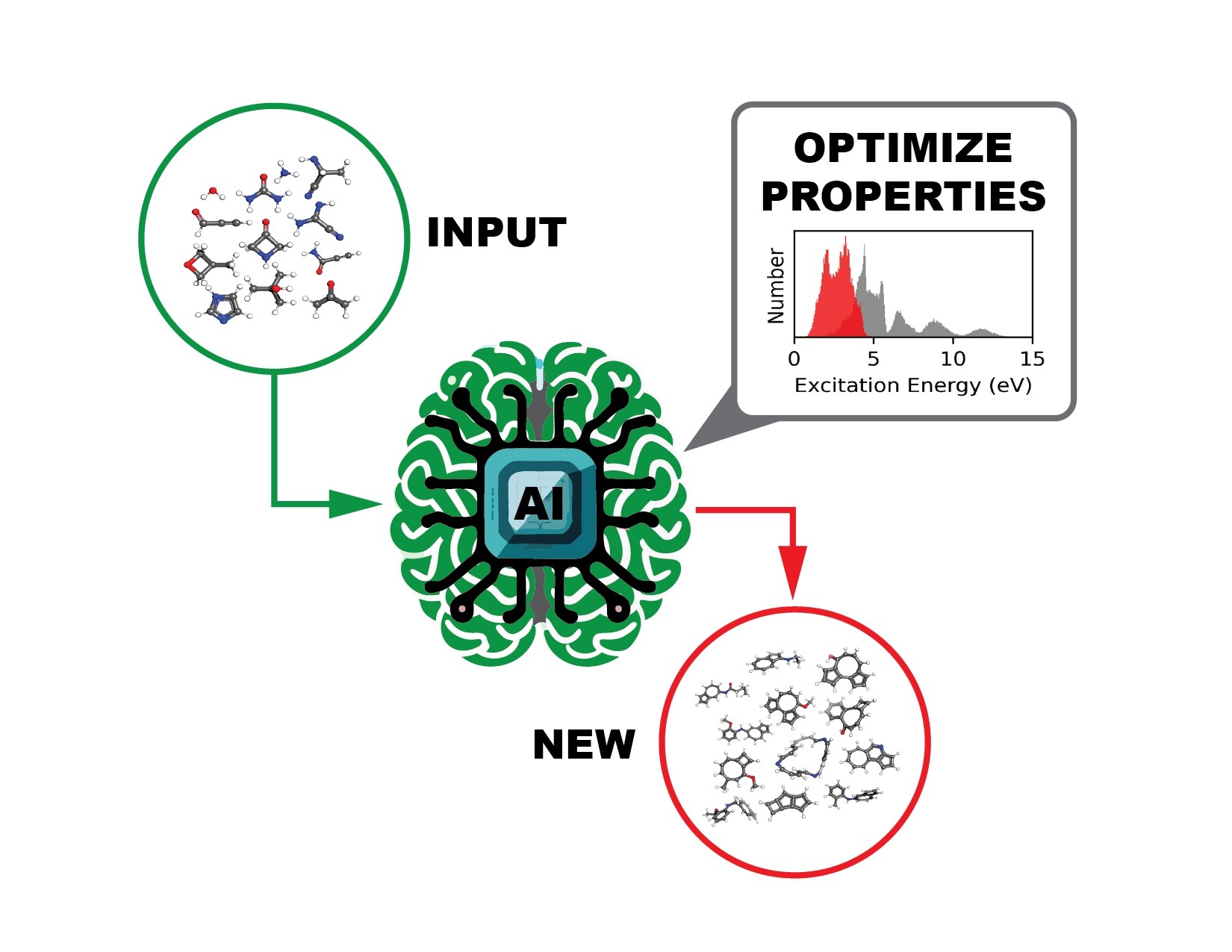
The AI agent, incorporating a language model-based molecular generator and a graph neural network-based molecular property predictor, processes a set of user-provided molecules (green) and produces/suggests new molecules (red) with desired chemical/physical properties (i.e. excitation energy). Image credit: Pilsun Yoo, Jason Smith/ORNL.
As a proof-of-concept test, the team generated the GDB-9-Ex dataset of 96,766 molecules composed of carbon, nitrogen, oxygen and fluorine, with at most nine nonhydrogen atoms. It showed that the designed workflow is effective and that the DL training accurately predicts the position and the intensity of the most relevant peaks of the ultraviolet-visible spectrum. From that initial success, the team ramped up its volume with the ORNL_AISD-Ex dataset, which contains 10,502,917 molecules composed of carbon, nitrogen, oxygen, fluorine and sulfur, with at most 71 nonhydrogen atoms. Pilsun Yoo, a postdoctoral research associate in Irle’s group, developed tools to analyze the resulting datasets.
The ultraviolet-visible spectrum, which describes a molecule’s excitation modes, was computed for each of the more than 10 million molecules. This information reveals what light frequency is required to target a molecule and break apart some bonds of the chemical compound. Another property of interest computed for each molecule was the HOMO-LUMO gap — the energy gap between the highest occupied molecular orbital and the lowest unoccupied molecular orbital — which reliably measures the molecule’s stability. With this information, a DL model could efficiently sift through the data to identify promising molecules for different prospective uses.
In fact, Lupo Pasini and his team at ORNL, including computational scientist in machine learning Pei Zhang and HPC data research scientist Jong Youl Choi, are developing just such a DL model: HydraGNN.
“The HydraGNN architecture takes in the atomic structure, converts it into a graph, and then it tries to predict as an output what the first-principles code would produce. It’s a surrogate model for expensive first-principles calculations,” Lupo Pasini said.
The results from HydraGNN’s training on the datasets, and its molecular discoveries, will be detailed in a forthcoming paper.
This research is sponsored by the Artificial Intelligence Initiative as part of the Laboratory Directed Research and Development program at ORNL. An award of computer time was provided by the OLCF Director’s Discretion program.
Related Publication:
Massimiliano Lupo Pasini, Kshitij Mehta, Pilsun Yoo and Stephan Irle, “Two excited-state datasets for quantum chemical UV-vis spectra of organic molecules,” Nature Scientific Data, August 2023. https://doi.org/10.1038/s41597-023-02408-4
UT-Battelle manages ORNL for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time. For more information, visit energy.gov/science.



