
Uncovering the molecular structures of proteins can help scientists elucidate biological functions of proteins and search for drug candidates for diseases. To better understand proteins, scientists can use imaging techniques to probe their molecular structures. However, many imaging techniques fall short of capturing details at the atomic level.
That’s why a team led by Abhishek Singharoy at Arizona State University is using the nation’s fastest supercomputer, the Oak Ridge Leadership Computing Facility’s (OLCF’s) IBM AC922 Summit, to model molecular structures in fine detail. The team recently simulated millions of structures and matched the results with experimental data to gain new insights into how proteins transition to different shapes, or conformations. The results, recently published in Matter, can point scientists to rare conformations of molecular structures.
“You often can’t see rare conformations with traditional modeling techniques,” Singharoy said. “But with Summit, we can capture these conformations by simulating ensembles of proteins.”
The team discovered rare conformations of Flpp3, a protein involved in tularemia (also known as “rabbit fever”), a bacterial disease that can be fatal. They found that in the most probable structure for the protein, it remains closed. However, in rarer conformations, the protein has a binding pocket that opens. This could give scientists a better understanding of how to thwart the disease process for tularemia.
Simulations such as these can also help scientists realize the flexibility of certain structures and capture proteins’ unfolding and symmetry breaking as they transition to different states.
Beyond experiment
Resolving the molecular architectures of biological entities like proteins, RNA, DNA, or macromolecules typically involves performing experiments to gain observational insights.
Scientists have a number of tools at their disposal to study molecular architectures experimentally. These include x-ray crystallography, the use of x-rays to detect crystal structures; nuclear magnetic resonance, in which a magnetic field is used to manipulate atomic nuclei in molecules to understand their structures; and cryo–electron microscopy, which involves freezing samples and using a beam of electrons to probe their molecular structures.
“Experimental techniques are useful to examine and determine the structures of molecular entities,” Singharoy said. “But unfortunately, there are experimental limitations, and scientists can’t often resolve these structures at the atomic level.”
Experimental limitations include the inability to fully capture the physical properties of the molecule or obtain a high-enough resolution. In biology, observation provides an average of molecular states rather than a snapshot of an instant.
“Observation isn’t exact—it’s an average,” Singharoy said. “There are different components of that average quantity that are hidden under the rug of the seen experimental dataset.”
The team earned an allocation of computing time on the OLCF’s Summit under the Innovative and Novel Computational Impact on Theory and Experiment, or INCITE, program to perform simulations and match them to observational data. This computing time enabled them to capture individual snapshots of proteins in different conformations rather than an average of their states. The OLCF is a US Department of Energy (DOE) Office of Science user facility located at DOE’s Oak Ridge National Laboratory.
“Summit’s parallel capabilities allowed us to unentangle averages using an ensemble representation,” Singharoy said. “Using Summit, we could tease out the rare structures that have implications in various biological processes.”
Revealing rare structures
The team used the nanoscale molecular dynamics code, or NAMD, combined with the open molecular modeling code, or OpenMM, to perform the simulations on Summit. Team members who ran the simulations included Mrinal Shekhar at the University of Illinois at Urbana-Champaign, Daipayan Sarkar at Purdue University and Arizona State University, and Alberto Perez at the University of Florida. Within NAMD, the team employed the molecular dynamics flexible fitting methodology, which fits modeling data to experimental data. Within OpenMM, the team used MELD (for modeling employing limited data). The team combined these codes into a pipeline they named CryoFold.
“This pipeline subsumes these two different methodologies,” Singharoy said.
The team investigated millions of structures and then matched them with experimental data. Summit’s parallel computing capabilities enabled the team to generate these structures at an unprecedented rate, Singharoy said.
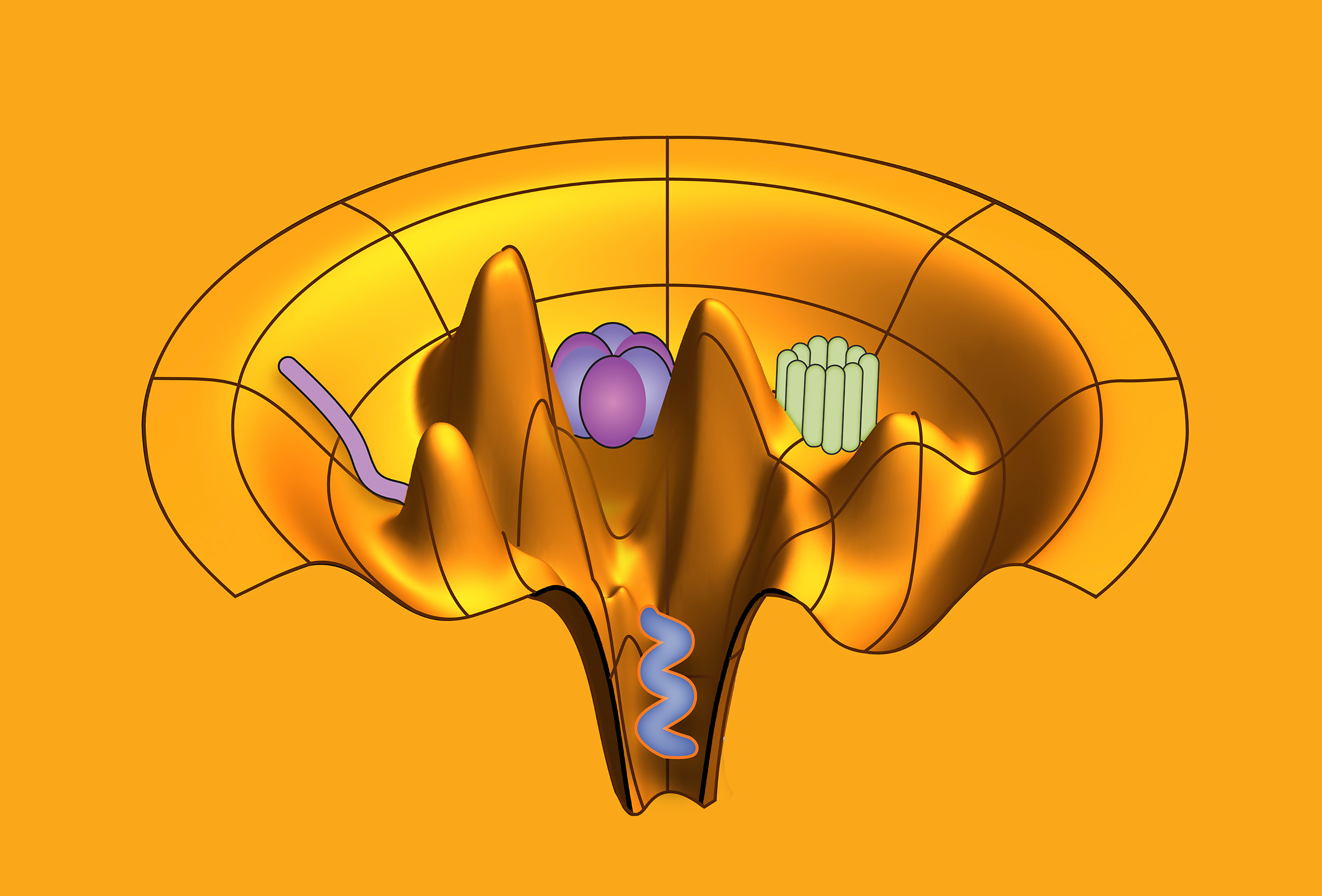
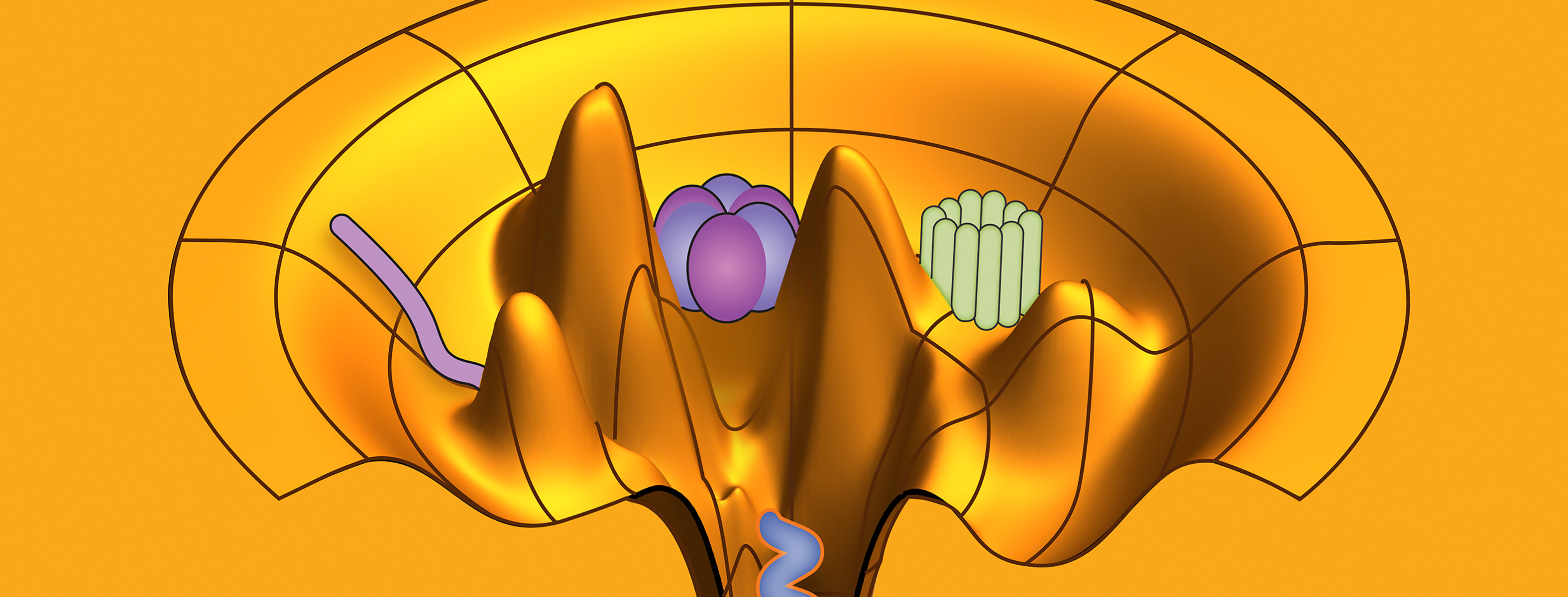
An ensemble refinement of molecular motors with CryoFold. Image Credit: Abhishek Singharoy, Arizona State University
One challenge that the team overcame was modeling a folding membrane.
“You need the presence of an explicit membrane model to fold membrane proteins, but CryoFold managed to do that, and we were able to extract info from the data. This hasn’t been done before,” Singharoy said.
The results can help scientists understand rare conformations and gain insights into the flexibility of biological systems. The team hopes to understand the flexibility of ATP synthase, a molecular motor that catalyzes ATP, the energy storage molecule.
“This is what ATP synthase is about,” Singharoy said. “We would’ve thought that ATP synthase is stiff, but it’s not. It’s like a hinge, and that hinge movement was hidden inside the data.”
Additionally, because proteins can exist in multiple conformations, scientists can gain understanding of a protein’s unfolding and symmetry breaking as it transitions to different states.
“When you’re looking at two end points, a change in shape might not have any bearing on the symmetry of the secondary structure. However, it might compromise the structure of the protein,” Singharoy said. “Biology might need the protein to transition at a particular rate, and for that to happen, the shape might need to break its symmetry. You wouldn’t be able to see these unfolded structures without this.”
AlphaFold and CryoFold coalesce
In future work, the team will investigate DNA and RNA complexes. Additionally, they are creating complementary tools, such as incorporating the AlphaFold protein structure database with CryoFold.
Thus far, the team has taken initial guesses from AlphaFold to construct data-guided protein ensembles.
“The idea of ensembles is still a bit far-flung for the structure-prediction piece,” Singharoy said. “But you can take the structure prediction piece and use CryoFold to make ensemble predictions.”
Related Publication: Mrinal Shekhar, et al., “CryoFold: Determining Protein Structures and Data-Guided Ensembles from Cryo-EM Density Maps,” Matter 4, no. 10 (2021): 3195–216, https://doi.org/10.1016/j.matt.2021.09.004.
UT-Battelle LLC manages Oak Ridge National Laboratory for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time. For more information, visit https://energy.gov/science.



