
One of the biggest challenges of drug discovery lies in the fact that scientists must search through an almost infinite space of chemical compounds to find one that might be capable of interfering with the infectious disease process. When the SARS-CoV-2 virus that causes COVID-19 began sweeping the globe, scientists knew that finding treatments would be no easy feat. The novel coronavirus was uncharted territory. Any protein–small molecule interaction might be the key to thwarting it.
A multi-institutional team, led by a group of investigators at the US Department of Energy’s (DOE’s) Oak Ridge National Laboratory (ORNL), has been studying various SARS-CoV-2 protein targets, including the virus’s main protease. This protein plays a key role in viral replication by snipping the virus’s newly made protein chain into smaller functional units that do the work to help it replicate.
Now, the team has harnessed 27,612 of Summit’s NVIDIA V100 GPUs to simulate more than one billion compounds binding with two different structures of the main protease, with each billion-compound screening completed in less than 24 hours. The feat has earned the team a finalist nomination for the Association of Computing Machinery (ACM) Gordon Bell Special Prize for High Performance Computing–Based COVID-19 Research, a special version of the ACM Gordon Bell Prize for outstanding achievements in high-performance computing to be presented at this year’s SC20 virtual conference.
The multi-institutional team was led by Ada Sedova, a biophysicist in the Molecular Biophysics Group within ORNL’s Biosciences Division; senior staff member Oscar Hernandez of ORNL’s Computer Science and Mathematics Division; computational scientists Josh Vermaas and Jens Glaser at the National Center for Computational Sciences (NCCS); and David Rogers, NCCS performance engineer.
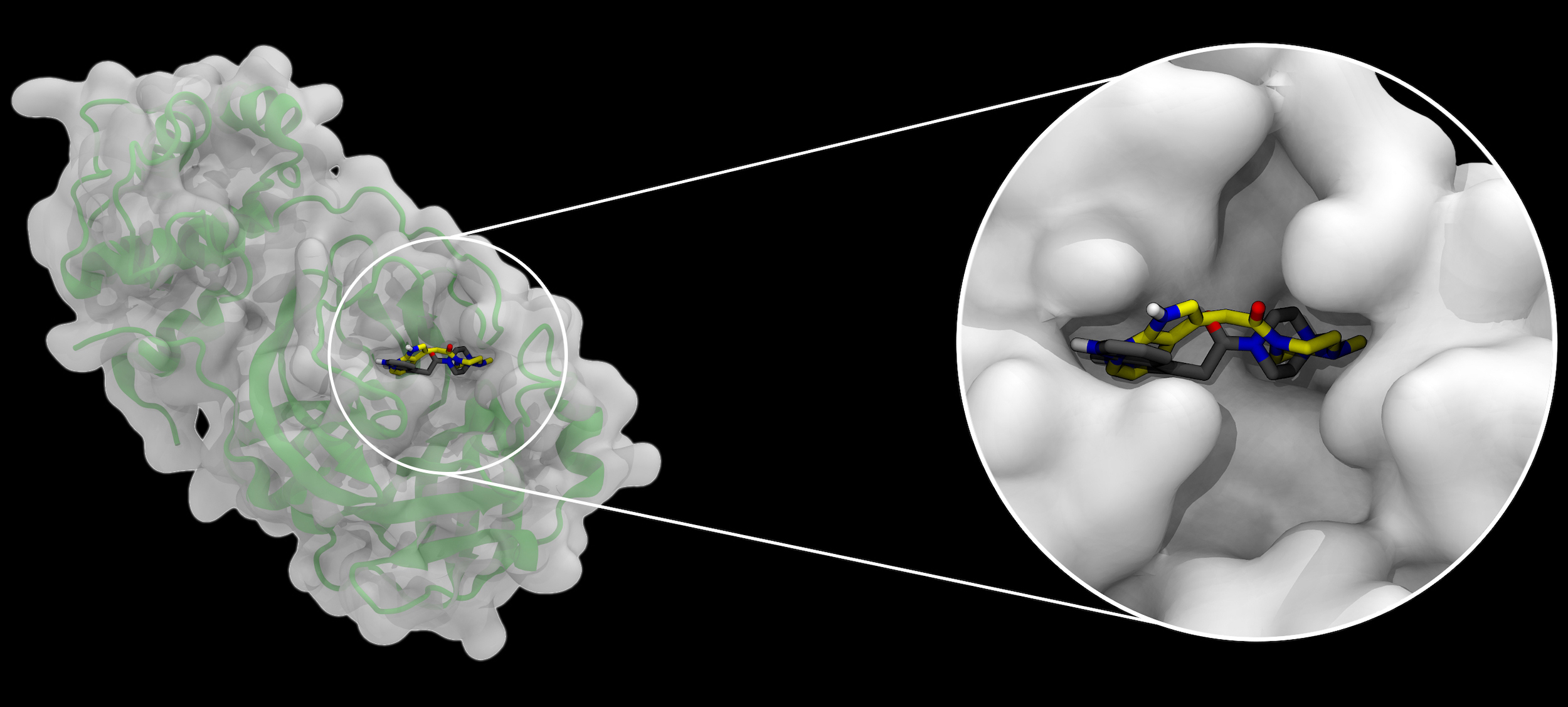
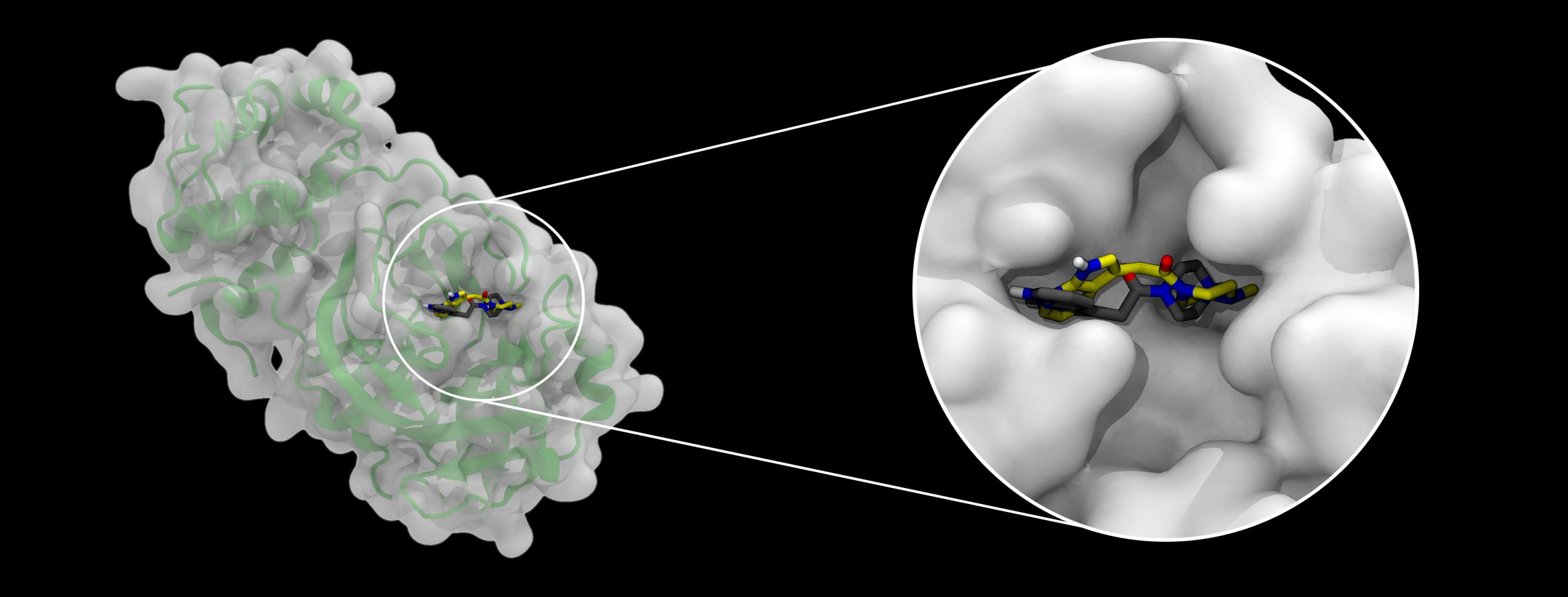
A docking example, emphasizing how binding pockets in a protein (left, with secondary structure shown as a green cartoon below a semitransparent surface), in this case the SARS-CoV-2 main protease, can be occupied by small molecules. Two alternative bound molecule configurations are shown, black representing a crystallographic structure, and yellow representing the pose predicted by AutoDock-GPU. Image Credit: Joshua Vermaas and Rachel McDowell, Oak Ridge National Laboratory
This project began earlier this year when these five lead investigators launched a collaborative effort with NVIDIA and Scripps Research to create and run a new version of the AutoDock-GPU molecular modeling code, optimizing it for high-throughput molecular docking simulations on the Summit supercomputer at the Oak Ridge Leadership Computing Facility (OLCF), a DOE Office of Science User Facility at ORNL.
The team—Sedova, Hernandez, Glaser, Vermaas, Rogers—worked with ORNL’s Swen Boehm, Matthew Baker, and Mathialakan Thavappiragasam; NVIDIA’s Jeff Larkin and Scott LeGrand; Andreas Tillack of the Forli Lab at Scripps Research; and Aaron Scheinberg of Jubilee Development to successfully scale AutoDock-GPU to Summit and carry out the massive screenings. The work was carried out using a portion of a special computational allocation awarded by the COVID-19 High Performance Computing Consortium.
The result was a 350-fold speedup over the CPU version of AutoDock-GPU, a 50-fold speedup per Summit node.
For each separate docking simulation, the team generated 20 possible poses, or configurations, showing how each synthetically producible compound might fit inside of the viral protein structure’s binding pocket. To accurately model the protein, the team used crystallographic structures from neutron scattering experiments performed at the High Flux Isotope Reactor and the Spallation Neutron Source at ORNL.
“When we were using Summit, we were docking 20,000 compounds a second,” Sedova said. “We have done this in 24 hours with full optimization of these poses, the way people would normally do at the small scale. To be able to do this on a billion compounds would have taken months on even the largest academic clusters without the optimizations of AutoDock-GPU for Summit.”
After generating the poses, the team had a lot of data analysis work left to do—a task that was led by Glaser, first author on the ACM Gordon Bell Special Prize finalist paper. The team calculated features of the poses to apply different machine-learning models to reevaluate them and to better determine whether or not each compound was a strong binder to the main protease.
“We also had to analyze the data efficiently to avoid thwarting the significant speedups we achieved in the docking phase,” Glaser said. “This is where Summit’s GPUs came to the rescue—again—and we used them to accelerate big data processing.”
To analyze the massive amount of data—1.3 terabytes per large-scale calculation—the team implemented a “virtual laboratory” on Summit to sort, manipulate, and join data pieces together. By employing the BlazingSQL platform, NVIDIA RAPIDS, the Numba compiler, and the Dask ecosystem to query data, the team reduced the processing of their results by orders of magnitude, achieving a 52-fold speedup for the full pipeline.
“We are accelerating the process of finding a successful molecule to suggest to experimentalists and ultimately to pharmaceutical companies to slow the spread of COVID-19,” Glaser said. “We are also exploring the chemical space of molecules rapidly so that we can make meaningful correlations and statistical predictions for the drug discovery process.”
The team’s results are being validated experimentally at ORNL by Liane B. Russell Distinguished Staff Fellow Stephanie Galanie, who is performing test-tube experiments of the main protease binding to different compounds. Based on her results, the researchers are refining their approach and informing future predictions. They also expect to look at other proteins in the future—including ones that may be even better targets for their computational approach.
The team is presenting their work at the SC20 conference tomorrow, November 19.
“We are still searching for the strongest binding molecules,” Sedova said. “We stood up a new computational capability that’s never been done before, and we’ve gained the ability to validate our approach. This work is providing us with a rich experimental data set that we can harvest and analyze to improve our results and ultimately get closer to our goal of finding the optimal molecules.”
This research was funded by a special COVID-19 Seed grant from ORNL’s Laboratory Directed Research and Development program.
Related Publication: Jens Glaser, Josh V. Vermaas, David M. Rogers, Jeff Larkin, Scott LeGrand, Swen Boehm, Matthew B. Baker, Aaron Scheinberg, Andreas F. Tillack, Mathialakan Thavappiragasam, Ada Sedova, and Oscar Hernandez. “High-Throughput Virtual Laboratory for Drug Discovery Using Massive Datasets.” To appear in International Journal of High Performance Computing Applications, 2020.
This team also acknowledges contributions from the following: Rupesh Agarwal, Jeremy Smith, and Micholas Smith of the University of Tennessee and ORNL; Omar Demerdash, Marti Head, Benjamín Hernandez, Robert Hettich, Jason Kincl, Dustin Leverman, Ketan Maheshwari, Stanton Martin, Don Maxwell, Julie Mitchell, Jeff Nichols, Ryan Prout, Arjun Shankar, Suhas Somnath, Spencer Ward, and Jack Wells, all of ORNL; Jess Woods of the Oak Ridge Institute for Science and Education; Joe Eaton, Peter Entschev, Geetika Gupta, John Kirkham, Jonathan Lefman, Joshua Patterson, Duncan Poole, and Zahra Ronaghi, all of NVIDIA; Stefano Forli and Diogo Santos-Martins of Scripps Research; Felipe Aramburu, Rodrigo Aramburu, William Malpica, and Shannon Smith, all of BlazingSQL, Inc.; Miles Euell, Jamie Kinney, and Usman Qureshi, all of Google; Andreas Koch and Leonardo Solis-Vasquez of TU Darmstadt; Hubertus van Dam and Shinjae Yoo of Berkeley National Laboratory; and J.C. Gumbart and the Gumbart Laboratory at the Georgia Institute of Technology.
UT-Battelle LLC manages Oak Ridge National Laboratory for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time. For more information, visit https://energy.gov/science.



