When Harel Weinstein and his team at the Joan & Sanford I. Weill Medical College (Weill Cornell Medicine) of Cornell University set out to learn the molecular mechanisms of drugs, they weren’t expecting to train computers to analyze some of the most complex data in pharmacology. In fact, they really weren’t expecting to train computers at all.
“Machine learning has never been used to classify these mechanisms for drug design—and certainly not at the scales we’re dealing with,” said Weinstein, Director of the Institute for Computational Biomedicine at Weill Cornell Medicine. “At the beginning of this study, the question was: how do you even present this data to a machine learning algorithm?”
Because of the complex nature of the data generated via the current method of modeling drug mechanisms, machine learning hasn’t been regarded as an ideal tool for analyzing it. The current method, called molecular dynamics (MD), simulates the movements and interactions of molecules in time and space, and the datasets generated contain large amounts of information about rapid movements that have little relevance when analyzing complex drug mechanisms.
When Weinstein’s PhD student, Ambrose Plante, was challenged to extract information from these MD datasets by using machine learning, he had to think first how to reconfigure MD data into a format tailor-made for machine learning. The way he achieved this? By transforming the information into a color picture that a machine learning algorithm could understand. With this transformation, a novel way of analyzing MD data was born—a way that might be the key to new discoveries in pharmacology.
Plante then set out to use machine learning as a tool to understand functional selectivity, which is the tendency for a drug (ligand) to bind to a particular kind of protein a cell membrane in a manner that causes the protein to select a certain signal transduction pathway inside. Other ligands binding to the same protein can modify it to select a different pathway and, thereby, a different function.
Understanding what change in the receptor triggers this preference is the key to uncovering the mechanism of functional selectivity and understanding how the hundreds of these G protein-coupled receptors (GPCRs) that are the main targets for 40 to 60 percent of all medications in existence today can be used for improving health.
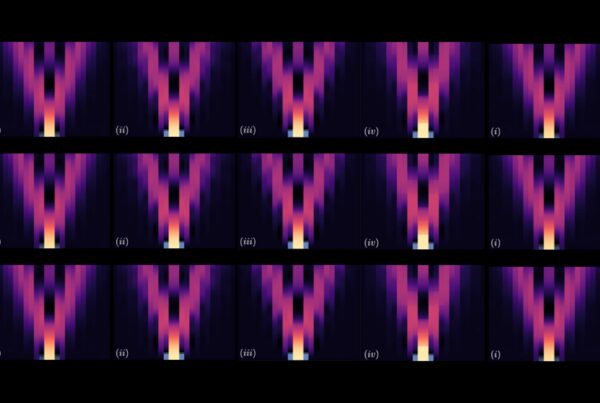
To complete a proof-of-principle experiment, the team used the most powerful supercomputer they could access—the 27-petaflop Cray XK7 Titan at the Oak Ridge Leadership Computing Facility (OLCF), a US Department of Energy (DOE) Office of Science User Facility located at DOE’s Oak Ridge National Laboratory. They turned 3D visual representations of MD data into 2D images for the first time to train an artificial neural network—a computing system that resembles the neural networks in the human brain—to determine how pharmacologically different classes of molecules either activate or inactivate certain signaling pathways in the brain.
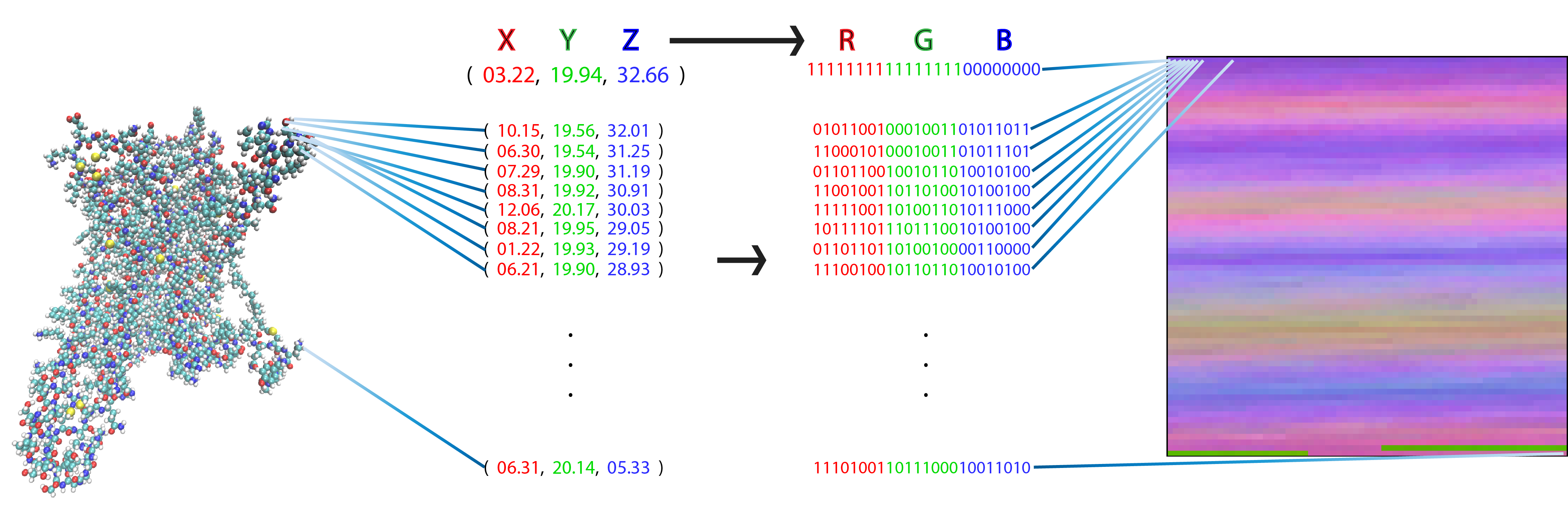
Each pixel in a flat, 2D image (right) represents one atom of a molecular figure (left) after transforming its 3D coordinates (X, Y, Z) into a red, green, blue (RGB) value. This particular representation has the special property that each pixel (i.e., matrix element) always represents the same atom in each frame from the trajectory of a particular protein. Image Credit: Harel Weinstein, Weill Cornell Medicine
The neural network the team created and trained was highly accurate in its determinations, and the team is already using it for even more complex problems.
The results of the GPCR study were published in 2019 in Molecules and were later highlighted by the editors of Trends in Pharmacological Sciences.
“These are the most popular receptors, in terms of the pharmacopeia,” Weinstein said. “The research is special because of the method by which these interactions were analyzed.”
Weinstein’s team currently uses the 200-petaflop IBM AC922 Summit supercomputer at the OLCF to understand the mechanisms of GPCRs and their signal transduction partners: the neurotransmitter transporters in the brain.
Bias in the brain
Communication in the brain occurs when signaling molecules are released by nerve cells and bind to receptors on other nerve cells through a process known as neurotransmission (“neuro” for “nerves” and “transmit” meaning “send, let through”). Signaling molecules called “neurotransmitters” serve this messaging system. These activate the receptors and are called agonists. Because neurotransmission is so important for life, nature has used neurotransmitter receptors—which were classified by scientists to characterize the GPCRs class of proteins—as targets in maintaining natural balances between prey and predators and has endowed some animals with molecules that block GPCRs to stop signaling by binding to them. These are called antagonists.
Following these clues from nature, drug designers developed drugs that mimic agonists, antagonists, and others (e.g., partial agonists) for those diseases in which GPCRs play major roles. Drug design efforts are thus geared toward gaining an understanding of how different molecules attain such pharmacological properties. This understanding would help scientists create drugs that are highly specific modulators of GPCR function, which work on only one kind of GPCR (and don’t affect others), and that don’t have effects other than the desirable ones (i.e., no side effects).
“The important thing is that you want what you’re designing to be selective—that it have action only at a particular type of GPCR,” Weinstein said.
Until 2013, scientists classified drugs by a system in which the drug’s action at a receptor determined its class. In the past few years, with the advent of new technology to analyze GPCRs, scientists have begun to discover that ligand interactions are much more complicated.
“It turns out that even though GPCRs typically signal through G proteins, there are competing signaling pathways to which an ‘activated’ GPCR can couple,” Weinstein said. The G proteins inside the cell—which receive and transmit information into the cell from the outside—compete with arrestin proteins. Both proteins want to couple to the activated receptor. Like its name suggests, if arrestin binds to the GPCR, it actually blocks the receptor from coupling to G proteins and sends a different message into the cell, causing vastly different responses.
But why would an activated GPCR that was considered to couple only to the G proteins (hence its name: G protein-coupled receptor) bind to arrestin instead?
The answer lies in the mysterious phenomenon of functional selectivity, or biased signaling. Although two drugs might activate the same receptor, the GPCR is a protein molecule that may be influenced differently by the two drugs to change its structure in different ways. Finding the differences in the GPCR’s responses and understanding the way ligand variations cause them are the goals of Weinstein’s team.
“To learn about this, we must do long simulations of the receptors bound to different ligands and follow their behavior in space and time,” Weinstein said. “We want to know which traits are specific to ligands that activate the receptors to bind arrestin rather than a G protein.”
Unlocking the details in 2D
The large amount of data necessary to understand the relevant receptor changes and discover which ligand traits are tied to this functional selectivity is a perfect problem for machine learning. But the machine has to ingest the information in a form that allows it to analyze them. A well-known form is a 2D image, but the drug-receptor complex is a dynamic 3D object.
The team first performed MD simulations using the Titan supercomputer at the OLCF after earning time on the system through an Innovative and Novel Computational Impact on Theory and Experiment, or INCITE, allocation.
Using this data, Plante took the 3D coordinates of each atom among the thousands of atoms in the GPCR-ligand complex and assigned each one a color representing its coordinate in the protein. Then, he assigned each atom to a pixel location and assembled the atoms into a 2D picture. He accomplished this for tens of millions of frames of the MD trajectory, turning each frame into a 2D picture-like representation, which resulted in a successful transformation without any loss of information from the trajectories.
The team then trained a convolutional neural network—which employs a sliding “window” to look at information—to classify agonists, antagonists, and inverse agonists that bind to specific neurotransmitter receptors in humans in the 5-HT2A and D2 receptors. These receptors are two of the most prevalent and important neurotransmitter receptors in humans, serving as targets for antidepressant and antipsychotic drugs, hormonal medications, and even psychoactive substances such as lysergic acid diethylamide (known by its common name, LSD).
The neural network achieved near-perfect accuracy in classifying the ligands bound to the receptor to their pharmacological class. For the 5-HT2A receptor, greater than 99 percent of the 2D frames analyzed by the neural network were correctly labeled.
After this task, Plante wanted to know what the neural network used to recognize the classes, so he employed an algorithm that determined how it made decisions. The analysis revealed the specific feature of receptor behavior that occurred only when a particular class of ligand was bound.
“When we look at the specific behaviors of the receptor molecule elicited by ligands in different classes, we can see that they involve very specific receptor regions,” Weinstein said. “The movements of residues in these regions differ when different ligands are bound, which helps us classify the ligands, but can also tell us something about the mechanism of the receptor—such as what an agonist does to activate a receptor. This study has really brought functional selectivity identification to a level of atomistic detail that never existed before.”
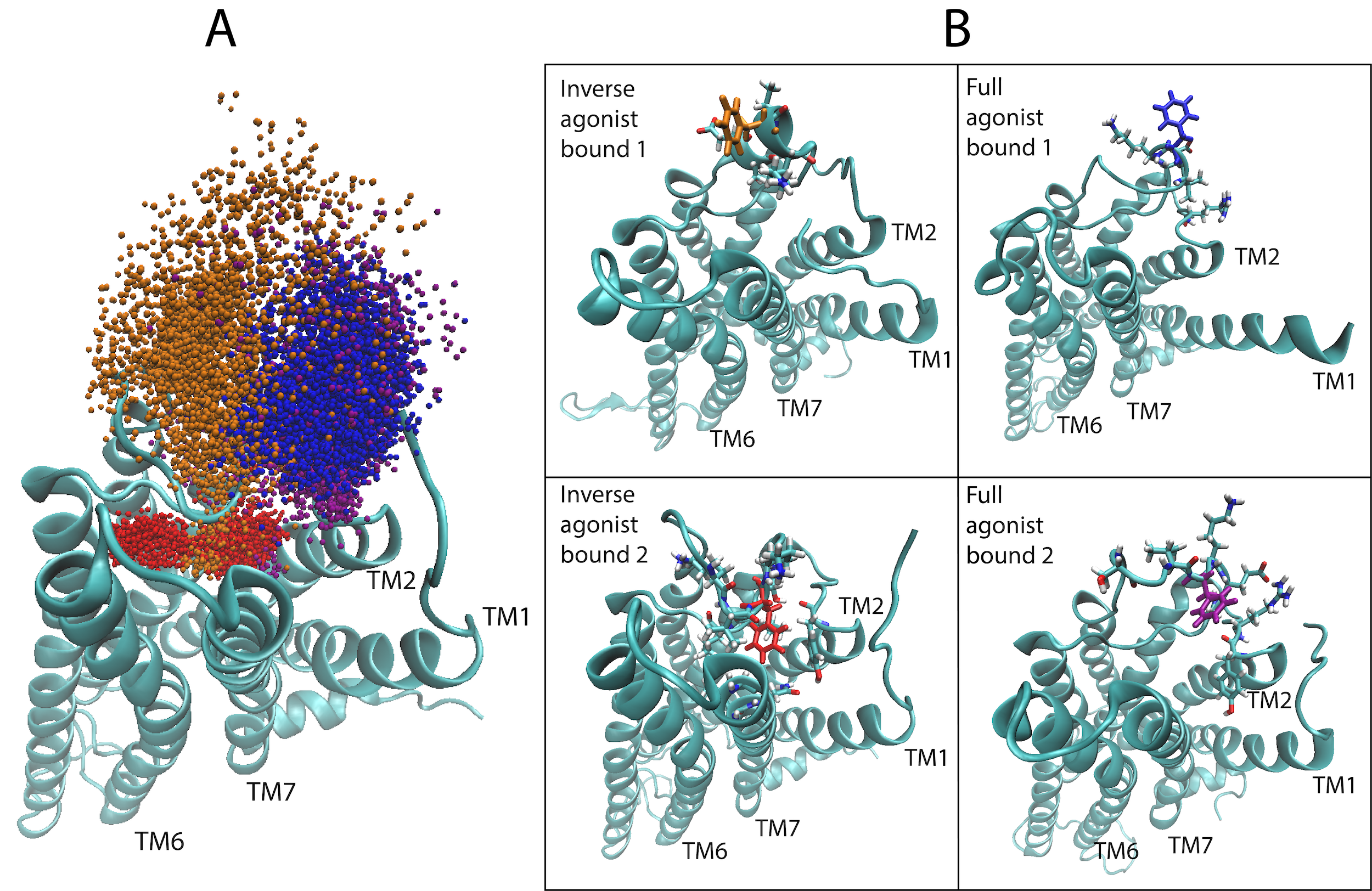
(Left) A depiction of the G Protein-Coupled Receptor (GPCR) 5-HT2AR (5-Hydroxytryptamine Receptor 2A), highlighting amino acid residue phenylalanine 222 (F222) that helps the protein function and responds to the binding of certain molecules (ligands). The machine learning algorithm showed that F222 responds in a ligand-dependent manner, favoring different locations or orientations when certain ligands bind but not others. This is illustrated here by the spatial sampling of the residue’s movements in the computational simulations of the 5-HT2AR bound to an inverse agonist ketanserin (red and orange)—which is a blood pressure drug—and the full agonist, the neurotransmitter serotonin (blue and purple). (Right) Frames from the trajectories of the inverse agonist and full agonist bound to the 5-HT2AR. Areas in which the protein spans the cell membrane are denoted transmembrane (TM). Image Credit: Harel Weinstein, Weill Cornell Medicine
The team said that because these specifically behaving regions are far away from the ligand binding site and are not easily seen to be central to the structural changes, they may not have been identified had it not been for the computing power at the OLCF.
A framework for future drug design
Weinstein’s team ultimately learned the path a certain ligand takes is what differentiates it from other ligands—even when the two bind to what appears to be the same place in the same receptor.
“Now, we know that if we want to design an agonist that will cause a similar response of the receptor, we have to design it so that when it binds with the receptor, it produces the same rearrangements,” Weinstein said.
One of the team’s major accomplishments is the transformation, for the first time, of MD simulation trajectories into images that can be recognized by machine learning technology. The team also found that changes in the receptor behavior are ligand-class specific (i.e., the neural network can recognize changes produced by agonists or inverse agonists, etc.), which builds a framework for further understanding the mechanisms of different GPCRs in humans. The same analysis framework is now used by the team to provide insight into how structurally similar ligands can bias the receptor’s response and produce different effects in the cell by eliciting different responses to their binding.
This framework could have implications for resolving problematic side effects of some ligands. For example, LSD (a psychoactive drug) causes hallucinations at doses in the microgram range by activating the 5HT2A receptor. Some psychiatric drugs can induce the phenomenon at the same receptor. However, not all ligands that activate this receptor induce hallucinations. Weinstein’s team believes machine learning could reveal the mechanisms that cause this dramatic difference between hallucinogenic and non-hallucinogenic drugs activating the receptor and thus, understand how certain unwanted phenomena can be eliminated. This would allow drug designers to create new drugs that don’t cause side effects or have addiction potential.
“Not only could this help us understand and potentially mitigate things like drug addiction, but it will allow us to look at drug design from a completely different point of view and with more specific and hence, more powerful criteria,” Weinstein said.
The entire classification method illustrated in the publication of this research also applies to other signaling proteins—not just the ones involved in GPCRs—in humans and other organisms. It also opens new opportunities for understanding processes such as neurotransmission and cell growth and proliferation (such as in cancer).
Related Publications: Ambrose Plante, Derek Shore, Giulia Morra, George Khelashvili, and Harel Weinstein, “A Machine Learning Approach for the Discovery of Ligand-Specific Functional Mechanisms of GPCRs,” Molecules 24, no. 11 (2019): 2097. doi:10.3390/molecules24112097.
Óscar Díaz, James Dalton, and Jesús Giraldo, “Artificial Intelligence: A Novel Approach for Drug Discovery,” Trends in Pharmacological Sciences 40, no. 8 (2019): 550–551. doi:10.1016/j.tips.2019.06.005.
UT-Battelle LLC manages Oak Ridge National Laboratory for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time. For more information, visit https://energy.gov/science.