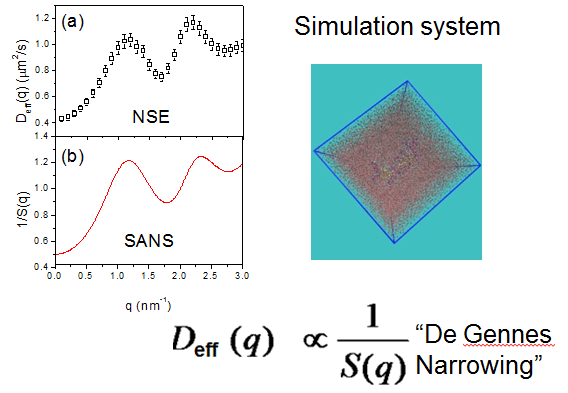
The great, Nobel-prize-winning theoretical physicist Pierre-Gilles de Gennes came up with a phenomenon known as “De Gennes Narrowing”, which relates the structure of a material to its motions. The structure and motion of the protein molecule can be characterized by Small-Angle Neutron Scattering (SANS) and Neutron Spin Echo (NSE) experiments, respectively. (Top right) A protein molecule embedded in a water box, simulated using a molecular dynamics approach.
Researchers use Titan, SNS to unravel the structure and function of one of nature’s most vital actors
The importance of proteins is difficult to overstate; they play a critical role in countless biological processes. An enhanced understanding of their structure and function is essential to advancing the state of the art in numerous arenas of science, from drug design to pollution mitigation to genetic engineering, but they are difficult to pin down.
Specific proteins serve different functions in various biological systems, and deciphering their individual roles is critical to understanding how they can best be used for scientific innovation. The first step in discovering their functions is to understand the motions of a protein’s domains—the distinct structures that all proteins possess and that researchers believe are vital to their purpose.
Deciphering the importance of these movements is inherently difficult because of the minute scales at which proteins operate. Traditional experimental techniques are either insufficient to explain them or prohibitively expensive, and because proteins change shape—or conformation—over time, a further timescale variable poses an additional obstacle to traditional observation.
Luckily for researchers, today’s fastest computers are capable of simulating these tiny marvels like never before, and few of today’s systems are faster than the Oak Ridge Leadership Computing Facility’s (OLCF’s) Titan. The OLCF is a US Department of Energy (DOE) Office of Science User Facility located at DOE’s Oak Ridge National Laboratory (ORNL).
The combination of Titan with ORNL’s Spallation Neutron Source (SNS), a DOE user facility that provides the most intense pulsed neutron beams in the world for scientific research and industrial development, offers researchers unprecedented tools for resolving one of molecular biology’s greatest mysteries: the ultimate function of protein domains.
That’s the strategy employed by Jeremy Smith, an ORNL/University of Tennessee Governor’s Chair who uses both Titan and SNS to understand the overall function and structure of proteins, research that was profiled in the April 18, 2014, issue of Physical Review Letters and the March 15, 2013, issue of Science.
Confirmation of experimentation
Most proteins consist of two domains, or “lumps,” as Smith calls them, which move relative to each other and help determine a specific protein’s function. Theoretically, if researchers can discover the relationship among a protein’s domain movements, the properties of that protein may more easily be revealed.
Using the neutron spin echo (NSE) spectrometer at SNS, a team of scientists performed two types of neutron scattering on protein samples: small-angle and ultrahigh-resolution spectroscopy. Subsequently Smith’s team used Titan to simulate protein dynamics using atomistic molecular dynamics and was able to show a relationship between the data collected in the SNS experiments.
The comparison of the neutron-scattering results demonstrated that the domain motion in a variety of proteins is inversely proportional to the interdomain structure factor, a quantity that characterizes the equilibrium spatial arrangement of the protein’s domains. The inverse dependence is qualitatively described by the de Gennes narrowing (DGN) theory, named for the famous French physicist Pierre-Gilles de Gennes, which posits an inverse relationship between the probability of a spatial configuration occurring and the fluctuation rate of the configuration—that is, a less populated configuration fluctuates faster.
Applying DGN to describe spectroscopic principles had previously been used only for structures like atoms, but Smith’s team showed it was applicable to proteins as well. The simulations were confirmed with further experiments at SNS, validating the team’s findings.
“Our biggest goal is to use supercomputing to understand neutron scattering,” said Smith, adding that “high-performance simulation is needed to interpret many of the experiments in the field.”
By showing a relationship between the two different types of neutron scattering via simulation, Smith’s team shed much-needed light on the function and shape of one of nature’s most vital components.
The mercury mystery
The project is part of a large research area that involves numerous teams from SNS, ORNL, the University of Georgia, and the Jülich Centre for Neutron Science and Institute for Complex Systems in Germany.
The team’s most recent work on protein motions is an extension of earlier research spanning multiple disciplines at ORNL that is focused on using bacteria to detoxify mercury-contaminated soil and water.
Methylmercury is toxic, and its toxicity works its way up the food chain, potentially poisoning entire ecosystems. Fortunately a specific set of bacteria contains a type of protein known as MerR, which possesses the unique property of detoxifying mercury. By performing a chemical reduction, these proteins render the methylmercury virtually harmless, making it a major weapon in ORNL’s quest for detoxification.
The bacterium is able to motor through methylmercury like a mini Pac-Man because it has a set of genes (containing the MerR protein) that looks like a metal staple and acts like a traffic cop. This regulator/cop first enables the mercury-processing bacterium to detect traces of the metal and then switches on genes in the bacterium’s DNA. Those genes are the blueprints for enzymes and other proteins that protect the bacterium from the toxic effects of methylmercury and can even help remove it from the microbe’s environment.
With this powerful arsenal of enzymes on hand, these bacteria can break down the highly toxic methylmercury to methane and mercuric mercury, which is subsequently turned into relatively harmless mercury vapor.
Smith uses a crab analogy to describe the process: “The claws grab mercury, and the mouth gets rid of it. The hands crawl all over the surface of the head to find the mouth.”
Titan and the NSE at SNS were critical throughout this research, and Smith’s team used the powerful duo to good effect when incorporating the results into a simulation.
“The supercomputer was used for the simulation of slow, large-scale molecular motions to interpret the data collected on the NSE instrument. The simulation of very long timescales requires a lot of processing power.”
The work has broad research implications.
“This work demonstrates that the combination of NSE with molecular dynamics code is useful for determining the nature of functional domain motions inside proteins,” said Smith. “It may presage wide application of these techniques in interpreting large-scale motions in biomolecules.”
Related publications
Liang Hong, Nikolai Smolin, and Jeremy C. Smith, “de Gennes Narrowing Describes the Relative Motion of Protein Domains.” Physical Review Letters 112 (April 18, 2014): 158102, https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.112.158102.
Jerry M. Parks et al., “The Genetic Basis for Bacterial Mercury Methylation.” Science 339, no. 6125 (February 7, 2013): 1332–1335, https://www.sciencemag.org/content/339/6125/1332.full.
Alexandre J. Poulain and Tamar Barkay, “Cracking the Mercury Methylation Code.” Science 339, no. 6125 (March 15, 2015): 1280–1281, https://www.sciencemag.org/content/339/6125/1280.full.
Oak Ridge National Laboratory is supported by the Office of Science of the US Department of Energy. The Office of Science is the single largest supporter of basic research in the physical sciences in the United States and is working to address some of the most pressing challenges of our time. For more information, please visit science.energy.gov