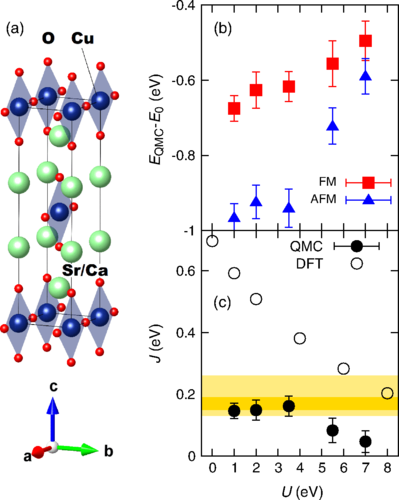
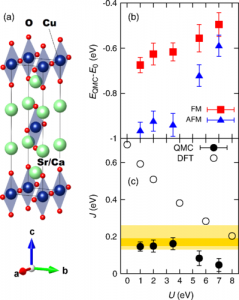
(a) The crystal structure of (Ca/Sr)2CuO3.
(b) The calculated quantum Monte Carlo (QMC) energies of different magnetic states. Lower QMC energies correspond to more accurate predictions. (c) The calculated mag-netic couplings from different methods, with the experimental range highlighted in yellow. The best QMC results from (b) give the best agreement with experiment, indicating that the QMC predictions are robust.
Image credit: Foyevtsova et a
First ab initio computational experiment of copper oxide moves scientists closer to understanding mysterious properties of cuprate superconductors
In 1911, Dutch scientist Heike Kamerlingh Onnes experimented with how mercury conducted electricity, and, in so doing, he opened a new avenue for study in materials science. Onnes found certain materials offered absolutely zero electrical resistance when cooled to a certain temperature. With this discovery, the superconductor era began.
Georg Bednorz and K. Alex Müller made a major leap in superconductors 75 years later by discovering a new set of materials that superconducted at much higher temperatures. These so-called high-temperature superconductors—almost always metal oxide-based materials—offer a future where power lines could send electricity through the grid with zero resistance. But 30 years after their discovery, scientists still do not know why these materials superconduct.
A group at the US Department of Energy’s (DOE’s) Oak Ridge National Laboratory wanted to better understand the complex interactions that enable superconductivity and needed one of the world’s fastest supercomputers to help them. Using the Cray XK7 Titan supercomputer at the Oak Ridge Leadership Computing Facility (OLCF), a DOE Office of Science User Facility, the team made its own leap in materials science research.
The group, led by ORNL researcher Paul Kent, advanced its understanding of superconducting by performing the first ab initio simulation of a high-temperature superconducting material, or cuprate.
DOE has long prioritized research into better understanding materials at the subatomic level and considers better understanding electron manipulation in novel materials one of its grand challenges. More specifically, by being able to better predict the behaviors of materials, scientists can accelerate research in a wide range of applications, including energy storage, catalysis, energy production, and metals that can be used as structural materials.
“The goal of this research was to calculate the so-called exchange coupling, or the magnetic interaction between adjacent copper atoms, from first principles,” Kent said. “The challenge is that metal oxides are very difficult to predict, and our method had never been applied to this class of materials before.”
Many materials scientists use density functional theory (DFT)—a computational method using mathematical functions to computationally model electrons—to try to model transition metal oxides accurately. Though DFT can offer accurate simulations, it has a difficult time simulating “d electrons,” which are more localized around atoms. To accurately simulate d electron-rich copper oxides, the team needed another method.
Kent’s team decided to use the quantum Monte Carlo (QMC) method for its simulations. Rather than using mathematical functions, QMC uses statistical data and random numbers to plot electrons in a simulation. Though QMC can be more computationally intensive work, it also allows the researchers to perform truly ab initio simulations far more accurately than DFT allows.
“When you get to transition metal simulations using DFT, they can start to make big qualitative errors,” Kent said. “Of course there are empirical fixes that you can do, but then of course you’re putting your hand into the simulation to fix the result based on knowing what you want to get. Fixing the result can be useful if you want to look at other properties, but it isn’t good if you want to make an ab initio prediction of magnetic couplings.” By using Titan, the Kent team was able to perform the first truly ab initio simulation of a cuprate.
Heavier atoms, harder simulations
After the team won an allocation through the Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program in 2013, it simulated calcium copper oxide for its first simulation. Kent explained the material choice had a lot to do with computational cost.
“This is one of the simplest materials that we could do that was still representative. It has a linear chain of copper oxide in it, rather than copper oxide planes,” Kent said, referring to the material’s one-dimensional molecular chains. “This is mainly for reasons of cost. We would like to do a lot more, so this is definitely a stepping-stone to doing more of the family of the cuprates. This is our proof of concept.”
One of the major challenges facing the team is memory. As atoms get heavier, the memory demands on the computer get larger. Most of the high temperature superconducting materials include heavy atoms. Accurately reproducing the behavior of the d electrons is particularly expensive
The team’s initial runs using its QMCPACK code were computationally expensive, but through the end of 2014, the team was able to reduce its memory usage by a factor of 8. This reduction allowed the team to do more computation on Titan’s GPUs and enabled computation on CPUs that previously required too much memory.
OLCF computational scientist Ying Wai Li helped the team use memory more efficiently and also created a new matrix inversion, allowing the team to delete hundreds of lines of source code, streamlining computational demands of running QMCPACK. Before Li’s work, the researchers were limited to around 730 electrons in a simulation. After she updated the code, the team was able to run simulations with over 1,000 electrons.
The team will continue to simulate heavier, more complex materials as computing resources allow. When the OLCF’s next-generation supercomputer, Summit, is finished in 2018, the team will be closer to simulating a broader range of materials, including heavier complex oxides for large-scale superconducting applications.
Related Publication:
K. Foyevtsova, et al. 2014. “Ab initio quantum Monte Carlo calculations of spin superexchange in cuprates: The benchmarking case for Ca2CuO3,” Physical Review X 4: 031003. doi: https://dx.doi.org/10.1103/PhysRevX.4.031003.
Image credit: Foyevtsova et al. Physical Review X. doi: https://dx.doi.org/10.1103/PhysRevX.4.031003
Oak Ridge National Laboratory is supported by the US Department of Energy’s Office of Science. The single largest support of basic research in the physical sciences in the United States, the Office of Science is working to address some of the most pressing challenges of our time. For more information, please visit science.energy.gov.