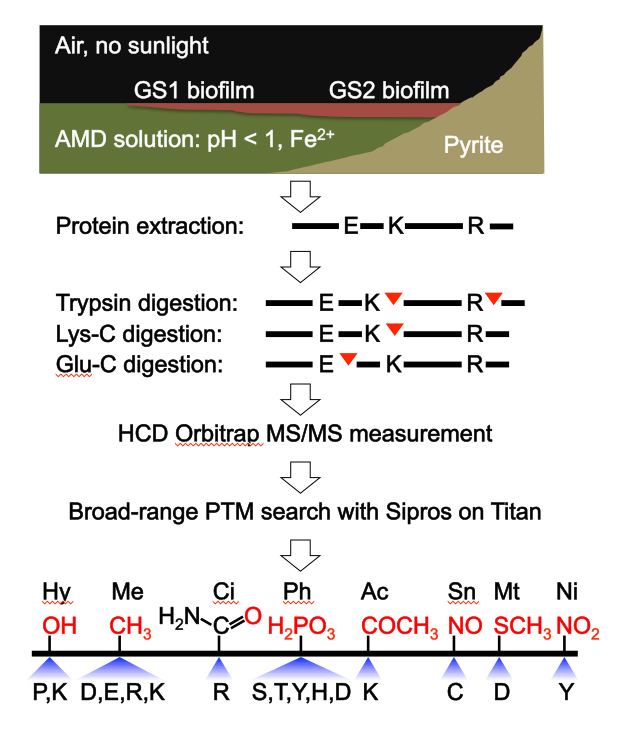
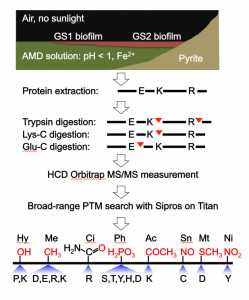
Acid mine drainage biofilms representing two growth stages (GS1 and GS2) were collected from a site within the Richmond Mine. Proteome samples were digested using three proteases in parallel and analyzed by high-energy collisional dissociation mass spectrometry. The researchers searched for various PTMs: hydroxylation (Hy), methylation (Me), citrullination (Ci), phosphorylation (Ph), acetylation (Ac), S-nitrosylation (Sn), methylthiolation (Mt) and nitration (Ni). The chemical formula (red) and modifiable amino acids are listed for each type of PTM. Image credit: Li et al.
First computational experiment of its kind sheds light on protein chemical changes that diversify microbes’ functions
Microbes are some of the world’s smallest organisms, but they play a big role in everything from the carbon and nitrogen cycles to environmental contamination.
Understanding microbial activities—and their response to climate change—is essential for understanding how climate shifts may affect large-scale systems, such as the carbon cycle. The US Department of Energy’s (DOE’s) Office of Science Biological and Environmental Research division has invested heavily in better understanding complex microbial processes.
This basic research need led a group of researchers at DOE’s Oak Ridge National Laboratory (ORNL) to explore the biological functions of microbial communities with high-performance computing. The team, led by ORNL researcher Chongle Pan, studies data from analyzing tiny organisms using mass spectrometry, an analytical chemistry technique. The team recently published the results of its computational analyses in Nature Communications.
“We combine high-performance computing with high-throughput biological measurements in our experiments,” Pan said. Specifically, University of California, Berkeley (UC Berkeley) biologists collected unique biological samples from the Richmond Mine in Iron Mountain, California. The ORNL researchers then measured these samples with mass spectrometry.
Mass spectrometry can generate millions of fragmentation patterns of positively or negatively charged molecules in a biological sample. Every fragmentation pattern contains thousands of unique data points. The ORNL researchers analyzed such complex, massive data using the Cray XK7 Titan supercomputer at the Oak Ridge Leadership Computing Facility (OLCF), a DOE Office of Science User Facility. The interdisciplinary team from ORNL and UC Berkeley extracted unique insights into protein chemical changes in natural communities at an unprecedented scale and resolution from the computational results.
Scientists know that microbes perform a wide variety of ecological functions, but they do not completely understand how microbes regulate those functions in different environmental conditions.
In fact, microbes drive a large portion of carbon cycling on Earth by fixing carbon dioxide in oceans and degrading terrestrial biomass such as falling leaves into carbon dioxide. The acid mine drainage system in Richmond Mine is a model carbon cycling system that includes microbial carbon fixation, which turns carbon dioxide into organic compounds, and degradation of submerged and decaying biomass back to carbon dioxide.
The team used a proteogenomics approach for studying microbes in this ecosystem. The genetic sequence of a microbe, or its genome, reveals the microbe’s potential functions. The changing protein makeup of a microbe, or its proteome, reveals its actual functions under specific growth conditions.
Often, proteins go through chemical changes called posttranslational modifications (PTMs) to diversify their functions. PTMs can alter, activate, or suppress the activities of proteins in response to different environmental conditions. However, because of the complexity of microbial communities, very little is known about how microbes use PTM in natural environments.
The team used a “shotgun,” or all at once, approach to address the problem of PTM identification; using high-performance computing, they searched many types of potential PTMs on all proteins encoded by microbes in their genomes. The team’s shotgun approach is computationally intensive work, but it offers a far more comprehensive picture of PTMs than those conventional approaches that experimentally enrich certain PTMs for study.
Without high-performance computing, Pan’s team would not have been able to use the shotgun approach to search through an astronomical number of modified protein sequences to identify many types of PTMs. The microbial communities in the Richmond Mine provided a glimpse into the diversity and dynamics of PTMs. This new proteomic approach revealed a wide variety of PTMs on many proteins. The identified PTMs greatly increased proteins’ structural and functional diversity.
In addition, the researchers found PTMs were substantially different between the early and late growth stages of the community, which suggests PTMs play a significant role on many microbes’ metabolic processes. Ultimately, the Pan team’s findings underscore the importance of PTMs in the physiology of microbial communities.
One of the major technical challenges facing Pan’s team was scaling the Sipros computational code—a database-searching algorithm that analyzes mass spectrometry data to identify PTMs—to Titan. The nation’s most powerful computer for open science, Titan is capable of 27 petaflops, or 27 quadrillion calculations per second. Pan’s lab received a Director’s Discretionary (DD) allocation in 2013 to scale Sipros up on Titan.
The team then received an allocation through the Advanced Scientific Computing Research (ASCR) Leadership Computing Challenge (ALCC) program in 2014. “We focused on our computational tools and development using the DD allocation, and then we used our ALCC allocation for full-scale analysis,” Pan said.
Pan credits his collaboration with OLCF support staff for getting Sipros scaled efficiently to Titan. Computer scientists at the OLCF were instrumental in helping the team effectively use Titan to run their simulations. In 2013, as part of the American Recovery and Reinvestment Act, the OLCF brought in a joint postdoc, Juanjuan Chai, to collaborate with Pan’s team. Chai played an integral role in scaling up Sipros using both the OpenMP and MPI interfaces to effectively use approximately 1,000 of Titan’s nodes—a very large number for proteomics applications. As computer architecture continues to change, Pan is confident that next-generation systems—such as the OLCF’s Summit supercomputer, set to be installed in 2018—will have the computational power for even larger-scale analyses.
“The next challenge is how to more effectively use GPUs on the machine. This process will only get easier on Summit because of architecture advances,” Pan said.
Related Publication:
Li, Z., et al. 2014. “Diverse and divergent protein post-translational modifications in two growth stages of a natural microbial community.” Nat. Commun. 5:4405. doi: 10.1038/ncomms5405.
Oak Ridge National Laboratory is supported by the US Department of Energy’s Office of Science. The single largest support of basic research in the physical sciences in the United States, the Office of Science is working to address some of the most pressing challenges of our time. For more information, please visit science.energy.gov.