Advances may lead to better catalysts and more efficient solar energy
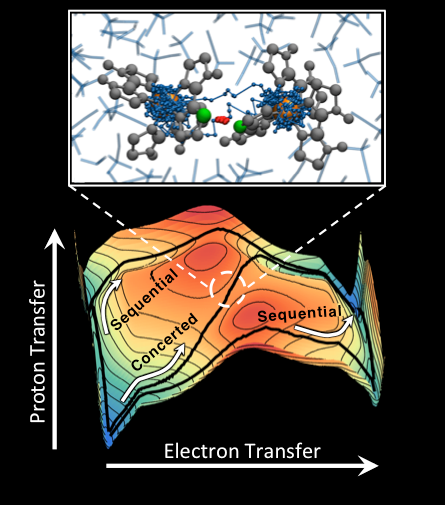
Below, the image shows competing sequential and concerted pathways for the PCET reaction in a symmetric iron bi-imidizole complex, with the reaction pathways overlaid on the energy landscape for the system in the ET and PT coordinates. Above, the image shows a snapshot of the molecular complex during concerted PCET, which illustrates the partially transferred electron (blue) and the partially transferred proton (red) in the ring-polymer molecular dynamics (RPMD) representation.
Plants solved the solar energy challenge billions of years ago, with photosynthesis.
The sunlight-fueled process begins with two very plentiful molecules—carbon dioxide (CO2) and water (H2O). By rearranging electrons, the plants are able to combine hydrogen, carbon, and oxygen to produce carbohydrates, the sugars that store all the energy needed to fuel the plants and, by extension, our own bodies.
Photosynthesis is impressive not only because plants found a way to convert sunlight into chemical energy that they could use. It is also impressive because they found a way to economize.
While transferring an electron alone is relatively hard, moving an electron along with a proton is relatively easy. Plants take advantage of this principle by moving the particles simultaneously. As a result they are able to rearrange more electrons, and produce more energy-storing carbohydrates, using less of the solar energy that drives the process.
This trick is known as proton-coupled electron transfer, or PCET. It is present in a variety of processes, not just photosynthesis, but we don’t really know how or why it works. The researchers who figure it out will open the door to a range of new and vastly improved technologies, from more efficient photovoltaics to more effective catalysts.
“Many times in a catalyst or solar cell it costs a lot of energy to just move an electron or proton,” explained theoretical chemist Thomas Miller of Caltech. “Finding ways to reduce that cost involves moving electrons and protons in a coupled fashion. Understanding that at a detailed level is a central challenge in the basic science of energy.”
Miller and colleagues are exploring the PCET puzzle through simulation on the OLCF’s Titan supercomputer. Titan is allowing the group to simulate PCET at a level that was previously impossible. The group discusses its work in recent issues of The Journal of Physical Chemistry Letters and The Journal of Chemical Physics.
“PCET is interesting for two reasons,” Miller explained. “First, it’s used a lot in nature, so it is clearly a useful strategy. And from a quantum mechanical perspective, it’s a tricky problem to understand—we need new theoretical methods to tackle it.”
PCET simulations are very complex and computationally very expensive. They must incorporate the behavior of quantum mechanical particles—electrons and protons in the case of photosynthesis—with that of particles obeying the rules of classical physics—for example, the surrounding water molecules.
Quantum particles are breathtakingly fast and seemingly bizarre; for example, they regularly cross solid barriers in a process known as quantum tunneling. Classical particles, on the other hand, are much slower and commonsensical; tunneling and other quantum behaviors are not available to them.
“In PCET you have to consider both very quantum mechanical electrons and weakly quantum mechanical protons,” Miller explained, “and you have hundreds of thousands of classical solvent molecules. How do you take that array of different behaviors and put it onto a reliable footing so you can describe what’s going on?”
Miller and colleagues have addressed this problem with a technique known as ring polymer molecular dynamics, or RPMD. Put simply, it is able to take the quantum mechanical behavior of electrons and protons and map them onto a simulation of classical molecular dynamics. The technique builds on the path integral formulation of Nobel laureate Richard Feynman, who also taught at Caltech.
The team has implemented RPMD within existing codes such as GROMACS (for Groningen Machine for Chemical Simulations) and DL_POLY. A typical simulation can include more than 14,000 atoms, including the reacting molecules and solvent environment.
In addition, it is able to work around the time differences among very fast quantum mechanical processes, slower classical behaviors, and the much slower reactions involved in PCET.
“These reactions can occur once per second or so,” Miller explained. “They can be very slow. If we did a direct molecular dynamics simulation, we would never see anything happen.
“So what we use are rare-event sampling strategies to efficiently sample only the reactive events and to overcome that very long timescale between reactive events, which can last up to seconds or minutes.”
Not only is this effort opening the door to new and improved technologies, but it also helped Miller secure tenure more than a year ahead of schedule.
One obvious application for the new understanding coming out of this work is improved technologies to use energy coming from the sun. Miller is working with two Caltech colleagues, experimentalists Harry Gray and Jonas Peters, to create photosynthesis without having to rely on plants.
“One of the things that people are trying to understand is how to make a purely synthetic device that does the job of photosynthesis efficiently.”
In particular, he said, the team is working to develop the ability to efficiently split water molecules to extract hydrogen. Success in this area could have significant impact on efforts to produce clean energy, because while hydrogen itself is a carbon-neutral source of practically limitless energy, it is now produced primarily from fossil fuels, which are not.
“There are experimentalists here at Caltech that are very focused on achieving the aim of finding catalysts that will split water to produce hydrogen and oxygen. To guide that effort, we need to understand the basic reactions that are governing that process.”
Related publication:
A. R. Menzeleev, J. S. Kretchmer, T. F. Miller, III, H. B. Gray, and J. M. Mayer, “Long-range proton-coupled electron-transfer reactions of bis(imidazole) iron tetraphenylporphyrins linked to benzoates,” Journal of Physical Chemistry Letters 4 (2013): 519−523.
J. S. Kretchmer and T. F. Miller III, “Direct simulation of proton-coupled electron transfer across multiple regimes,” Journal of Chemical Physics 138 (2013): 134109.