
Alloys—mixtures of two or more metals—are designed to be stronger, more ductile (pliable), or more durable than pure elements. Steel is one of the most popular alloy materials, but its weight makes it less desirable than lightweight alloys, which are favored in engineering for their ability to improve vehicle efficiency and reduce emissions.
Magnesium forms the lightest alloy known today, but its main drawback is its brittleness—a property that leads to cracking. Scientists aim to understand small defects called dislocations that contribute to this cracking because mitigation strategies depend on knowledge of these structural defects.
A team led by Vikram Gavini, associate professor of mechanical engineering at the University of Michigan, used the 200-petaflop IBM AC922 Summit supercomputer at the Oak Ridge Leadership Computing Facility (OLCF) to perform a simulation of a 10,000-atom magnesium dislocation system, a feat that could allow scientists to understand which alloying materials to add to improve magnesium’s ductility. By simulating a system tenfold larger than was possible with previous codes and 20 times faster, the team ultimately achieved a 200-fold speedup, running at 46 petaflops and earning an Association for Computing Machinery Gordon Bell Prize finalist nomination.
“Using Summit, we were able to perform the largest simulation to date of this dislocation system in magnesium that we believe is crucial to improving its ductility,” Gavini said.
If scientists can simulate alloys with high accuracy, they will gain the ability to prescreen new materials before experimental testing, thus accelerating the introduction of new materials. And with knowledge of the energetic principles involved in certain types of dislocations, experimentalists can more easily develop recommendations for new alloys.
Once they are in production, lightweight alloys could have impacts in fields ranging from transportation to aerospace.
Accommodating atomic shifts
To create alloys, materials scientists called metallurgists mix metals in a molten state and then pour the resulting liquid into a mold where it solidifies. As it hardens, dislocations sneak in, spreading throughout the material. When an alloy is bent or deformed, it can either accommodate the deformation by dislocation motion (a property known as ductility) or break, depending on the way its atoms are structured.
Ductile materials, like aluminum, have a cubic structure in which atomic shifts can result in dislocations that move in multiple directions and accommodate deformation. Other materials, such as magnesium, have a stricter structure. With limited pathways for the dislocations to move, the material must break to accommodate the applied deformation.
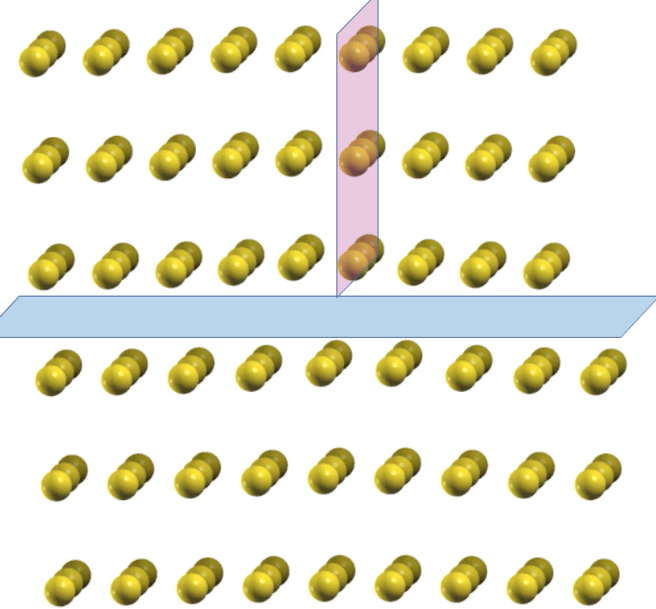
An animation of a dislocation moving through a system. The dislocation (pink plane of atoms) easily glides through the system along the blue plane. Image Credit: Vikram Gavini, University of Michigan
“In materials like magnesium, the dislocation activity is primarily on one plane, but in materials that have cubic structures, the dislocations can glide on many different planes,” Gavini said. “The idea is that, in magnesium, if we can get the dislocations to glide on new planes by adding new elements, this could prevent cracking.”
To understand materials, Gavini’s team performs density functional theory (DFT) calculations, which focus on the electronic structure of materials systems. Until now, existing DFT codes have been incapable of handling the large system sizes needed to accurately model these defects. To address these limitations, Gavini’s research group has, over the years, developed methods and algorithms for fast, accurate, and scalable DFT calculations.
Last year team members attended an OLCF hackathon, a hands-on workshop during which researchers port their existing codes to GPUs with the help of expert mentors. Before the hackathon, they had no existing GPU code, but at the event they were able to move their code onto Summit, the world’s most powerful and smartest supercomputer for open science.
“All of our algorithms were stable on CPUs, but we knew we had to have a GPU-based code to fully take advantage of an architecture like Summit’s,” Gavini said of the system at the OLCF, a US Department of Energy (DOE) Office of Science User Facility located at DOE’s Oak Ridge National Laboratory.
The team spent 6 months porting 90 percent of its code, DFT-FE (DFT with Finite Elements), to GPUs after earning time on Summit through a Director’s Discretionary allocation.
The team performed smaller simulations of magnesium dislocation systems with around 2,000 atoms on Summit before performing a final 10,000-atom simulation. At 13.6 nanometers by 13.6 nanometers by 1.2 nanometers, this final simulation provided the team with the accuracy needed to understand the energetic properties of the magnesium system at high fidelity. Information gleaned from these simulations can guide experimentalists as they add new elements to magnesium alloys to induce more “deformability” and reduce the possibility of cracking.
“This is the result of a culmination of work over the past 7 years,” Gavini said. “It’s good to be where we are.”
Spanning many fields
The team hopes the GPU-accelerated version of the code will serve as a computational tool for scientists to prescreen and identify crucial alloys that experimentalists could further refine. Continued refinements, Gavini said, could speed up alloy development across the industry.
The team also believes these developments could have applications in a variety of fields. Although the team demonstrated the code’s capability using a magnesium alloy, the researchers are confident it can be used for numerous problems of scientific interest.
“People want to understand how different nanoparticles could accelerate or catalyze certain chemical reactions, and for this, one needs large-scale DFT calculations that can be done quickly,” Gavini said. “This code could be used for that.”
Additionally, the code could be used to study the underpinnings of quantum transport in materials, the behavior of biological molecules, or the electronic structure of materials besides alloys.
“This is just one important scientific problem that we chose to demonstrate this code’s relevance, but it can be used for many other kinds of calculations,” Gavini said.
The work was supported by the DOE Office of Science through the PRISMS (Predictive Integrated Structural Materials Science) Center at the University of Michigan.
Related Publication: S. Das, P. Motamarri, V. Gavini, B. Turcksin, Y. W. Li, and B. Leback, “Fast, Scalable and Accurate Finite-Element Based Ab Initio Calculations Using Mixed Precision Computing: 46 PFLOPS Simulation of a Metallic Dislocation System.” To appear in SC19 Proceedings of the International Conference for High Performance Computing, Networking, Storage, and Analysis, Denver, CO, November 17–22, 2019.
UT-Battelle LLC manages Oak Ridge National Laboratory for DOE’s Office of Science, the single largest supporter of basic research in the physical sciences in the United States. DOE’s Office of Science is working to address some of the most pressing challenges of our time. For more information, visit https://energy.gov/science.



