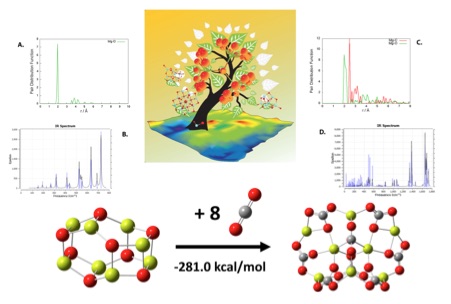
Bottom: (MgO)8 cluster reacting with eight molecular equivalents of CO2 to form an (MgCO3)8 cluster. Top mid: Graphical description for the TG-HGA method. A: calculated pair distribution function vs. radius plot for (MgO)8. B: calculated infrared spectrum for (MgO)8. C: pair distribution function vs. radius plot for (MgCO3)8. D: calculated infrared spectrum for (MgCO3)8.
Multi-organization project uses tree growth algorithm to simulate metal oxide cluster behavior
Postdoctoral researcher Mingyang Chen earned his doctoral degree at the University of Alabama, working closely with his mentor, David Dixon, in using computation to understand metal oxide clusters’ mysterious behaviors. Once he started working at the US Department of Energy’s (DOE’s) Oak Ridge National Laboratory in 2014, he continued collaborating with his old team, and the teamwork is paying off.
Chen now works in the scientific computing group at the Oak Ridge Leadership Computing Facility (OLCF), a DOE Office of Science User Facility, and has significantly contributed to how Dixon’s team approaches these complex materials problems computationally.
As he finished his doctoral work, Chen was tasked with developing an algorithm to understand metal oxide clusters better. Clusters—pre-nucleation clusters, to be more specific—are groups of molecules and nanostructures that begin to interact and form crystals but have not yet formed a nucleus, so they are not considered cells.
Certain clusters, such as the magnesium carbonate material the team most recently simulated, may be useful in preventing carbon emissions from polluting the atmosphere but do not appear naturally. Further, creating them in a laboratory is an expensive, difficult process.
Chen developed the tree growth–hybrid genetic algorithm (TG-HGA) in hopes of simulating how particles form clusters and how cluster properties evolve as the size increases and surface-to-volume ratio changes. The team first tried simulating a two-element cluster as proof of concept, and the titanium oxide simulations turned out to line up closely with experimental data.
After Chen moved to the OLCF and began using the Titan supercomputer—a Cray XK7 capable of 27 petaflops, or 27 quadrillion calculations per second—the team thought it could try simulating a more complex material. Unlike titanium–oxygen-based titanium oxide, magnesium carbonate consists of magnesium, carbon, and oxygen atoms. The extra element affects the degrees of freedom in a calculation, making it far more computationally intensive.
The algorithm still worked well with the more complex cluster, and the team—which also includes Andrew Felmy of Pacific Northwest National Laboratory and Virgil Jackson from the University of Alabama—was able to simulate magnesium carbonate clusters accurately and, ultimately, began to see how they grow into larger, more complex bulk solids. The team published its findings in the Journal of Physical Chemistry.
The team’s next set of simulations deals with cluster reactions to small molecules such as water and acidic gases, adding yet another layer of complexity to the project. The team is optimizing its algorithm further in hopes of not only simulating more complex clusters and their interactions, but also modeling how cluster particles interact with various surfaces or how clusters may react when exposed to light.
Chen noted that the main limitations do not relate to computational capabilities as much as actual scientific challenges. “These simulations are difficult, but the bottleneck is not scaling up the program,” he said. “The bottleneck deals with what happens when clusters get larger. It gets more and more difficult to substruct the correct genomes and put them together. That’s a scientific difficulty.”
Related Publication: M.Chen, V. Jackson, A. Felmy, D. Dixon. The Journal of Physical Chemistry. 119. 3419-3428 (2015).
Oak Ridge National Laboratory is supported by the US Department of Energy’s Office of Science. The single largest supporter of basic research in the physical sciences in the United States, the Office of Science is working to address some of the most pressing challenges of our time. For more information, please visit science.energy.gov.